Article Text

Abstract
Background and aim Martin–Probst syndrome (MPS) is a rare X-linked disorder characterised by deafness, cognitive impairment, short stature and distinct craniofacial dysmorphisms, among other features. The authors sought to identify the causative mutation for MPS.
Methods and results Massively parallel sequencing in two affected, related male subjects with MPS identified a RAB40AL (also called RLGP) missense mutation (chrX:102,079,078-102,079,079AC→GA p.D59G; hg18). RAB40AL encodes a small Ras-like GTPase protein with one suppressor of cytokine signalling box. The p.D59G variant is located in a highly conserved region of the GTPase domain between β-2 and β-3 strands. Using RT-PCR, the authors show that RAB40AL is expressed in human fetal and adult brain and kidney, and adult lung, heart, liver and skeletal muscle. RAB40AL appears to be a primate innovation, with no orthologues found in mouse, Xenopus or zebrafish. Western analysis and fluorescence microscopy of GFP-tagged RAB40AL constructs from transiently transfected COS7 cells show that the D59G missense change renders RAB40AL unstable and disrupts its cytoplasmic localisation.
Conclusions This is the first study to show that mutation of RAB40AL is associated with a human disorder. Identification of RAB40AL as the gene mutated in MPS allows for further investigations into the molecular mechanism(s) of RAB40AL and its roles in diverse processes such as cognition, hearing and skeletal development.
- Genetics
- guidelines
- molecular genetics
- clinical genetics
- microarray
- copy-number
- complex traits
- developmental
- epigenetics
- academic medicine
- genome-wide
- genetic epidemiology
- chromosomal
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Genetics
- guidelines
- molecular genetics
- clinical genetics
- microarray
- copy-number
- complex traits
- developmental
- epigenetics
- academic medicine
- genome-wide
- genetic epidemiology
- chromosomal
Introduction
Mutations in genes encoding Ras proteins are responsible for a number of human genetic disorders, including Costello (HRAS), Noonan (KRAS and NRAS), cardiofaciocutaneous (KRAS) and autoimmune lymphoproliferative (NRAS) syndromes.1 2 Ras proteins are components of the Ras/mitogen-activated protein kinase pathway and are important for proper cellular growth, differentiation and proliferation. Ras proteins canonically cycle (or switch) between active guanosine triphosphate (GTP)-bound and inactive guanosine diphosphate (GDP)-bound conformations (Ras-GTP and Ras-GDP).3 Ras proteins generate distinct signal outputs in cells, despite interacting with a common set of competing activators (GTPase activating proteins) and exchange factors (guanine nucleotide exchange factors) which regulate Ras-GTP levels.4 5 Ras proteins contain a hypervariable (HVR) domain within the C-terminal 25–50 amino acids. The HVR contains sites for post-translational lipid and other modifications and is the only region that differs significantly in sequence among the otherwise highly homologous Ras genes. HVR domains confer distinct biochemical properties of the Ras proteins and are involved in subcellular localisation and signal transduction. Cell- and tissue-specific expression may also contribute to the unique biological and developmental effects of Ras proteins.6
Rab GTPases belong to the Ras superfamily of small GTPases.7 More than 50 Rab proteins have been described in mammalian cells, each with a specific subcellular localisation and many with specific patterns of tissue distribution.8 9 Rab proteins are involved in regulating intracellular vesicle transport and trafficking of proteins between organelles, behaving as membrane-associated molecular switches.8 9 The suppressor of cytokine signalling (SOCS) box distinguishes the four Rab40 proteins (Rab40a, Rab40al, Rab40b and Rab40c) from other Rab GTPases. SOCS box-containing proteins interact with an E3 ubiquitin ligase complex (with elongins B and C) that polyubiquitinates target proteins for subsequent degradation.10 The target specificity of the complex is determined by a protein–protein interaction domain located N-terminal of the SOCS box-containing protein.10 RAB40AL is a 278-amino acid Ras-like GTPase protein with a single SOCS box immediately proximal to the C-terminal HVR domain.
RAB40AL (RLGP) was previously implicated in a male person with an inversion (46,X,inv(X)(p21.2q22.2)) disrupting the RAB40AL promoter region.11 This individual was diagnosed with congenital muscular dystrophy accompanied by severe intellectual disability, congenital nystagmus and athetosis. He never developed meaningful speech nor sat without support, and died at 18 years of age from suffocation due to tracheal obstruction. He was also diagnosed with congenital Duchenne-type muscular dystrophy and harboured a deletion of DMD exons 43–60, but his severe intellectual disability was ascribed to the disruption of RAB40AL function.11 The authors hypothesised that RAB40AL may play a critical role in the development or function of the central nervous system (CNS) by transducing signals or transporting molecules across mitochondrial membranes.11
Martin–Probst syndrome (MPS; MIM 300519) is a rare multi-organ system neurodevelopmental disorder initially characterised in three related male subjects with sensorineural hearing loss, cognitive impairment, short stature and craniofacial dysmorphisms.12 MPS shares some characteristics with Costello syndrome (CS) and Noonan syndrome (NS) such as short stature, cognitive impairment and craniofacial dysmorphisms. However, unlike CS and NS, individuals with MPS also exhibit sensorineural hearing loss, renal insufficiency and impaired haematopoiesis.12 13 Additional clinical features seen in one individual with MPS (patient 1 in Martin et al,12 same as individual II-1 in figure 1A) include chronic dyspnoea, pulmonary hypertension, septal cirrhosis and abnormal branching patterns of large hepatic vessels (unpublished data). The causative mutation for MPS was previously localised to a 68 megabase common haplotype region on X chromosome containing 683 genes between DXS1003 and DXS1220 (chrX:46,419,359-114,514,483; hg18; Xp11.3-Xq23).12 13 Deletions >1kb were excluded by PCR.12 13 Candidate genes COL4A5, DIAPH2, POU3F4, ATRX, TIMM8A, XNP, ALEX2, KCNE1L and UBE1, located within the 68 megabase target region, were previously excluded by direct sequencing or dHPLC.12 13 Female carriers in the pedigree have been found to exhibit non-random (or skewed) X-inactivation.13 Here we provide evidence that a germline missense mutation in RAB40AL leads to MPS.

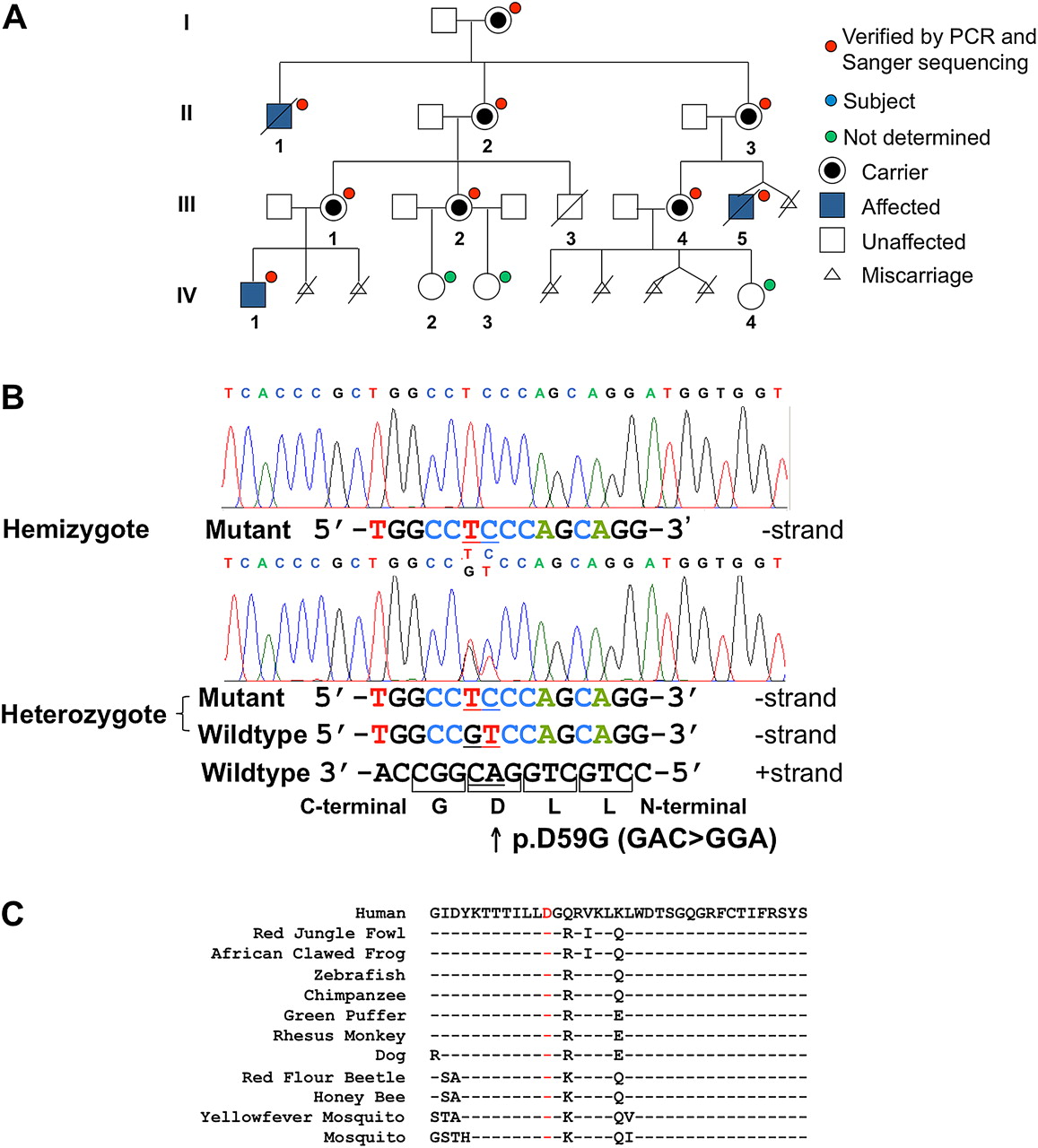
RAB40AL p.D59G variant analysis and segregation. (A) The p.D59G variant identified in the sequenced individuals (III-5 and IV-1) segregates with the phenotype in the family. For individuals with a red dot, p.D59G was identified by Sanger sequencing. Affected individuals are shown in blue. For individuals with a green dot, p.D59G status was not determined. (B) Sequence chromatograms showing the two consecutive nucleotide changes (AC→GA) in a male subject (hemizygous) affected with MPS and an unaffected obligate female carrier (heterozygous). This change results in an Asp to Gly change at codon 59. Sanger sequencing readouts are shown. (C) The p.D59G variant (red) lies within a highly conserved 37 amino acid region of the GTPase domain and is conserved from humans to mosquitoes.
Materials and methods
DNA samples from individuals were obtained with informed consent as approved by the Institutional Review Board for Human Subject Research at the University of Michigan Medical Center.
Variant discovery and analysis via massively parallel sequencing
We performed whole genome sequencing (WGS), whole exome sequencing (WES) and X chromosome-specific exome14 15 sequencing to identify the causative mutation in MPS (see Supplemental Methods for detailed methods). It should be acknowledged that the word ‘whole’, used here in the context of massively parallel sequencing, does not imply absolute and complete coverage of all human genomic or exonic sequences. Coverage is limited by the repetitive nature and %GC content of the target genomic regions and by the actual probes used in the various exonic capture methods or reagents used. Reads from all three sequencing strategies were aligned to the reference human genome (UCSC hg18) using BWA.16 For whole exome and whole genome data, SAMTools17 was used to remove duplicate reads and call single nucleotide variants (SNVs) and indels. For X chromosome exome data, duplicate sequences were removed using PICARD and quality scores were recalibrated and variants called using GATK.18 SNVs with a Phred quality score ≤30 or <4× coverage were excluded. SNVs were filtered through dbSNP130 and then imported into SeattleSeq for variant annotation. Variants were further filtered to exclude those identified by the 1000 Genomes Project (March 2010 release) or the National Heart, Lung, and Blood Institute Exome Sequencing Project. Predictions of functional effect of the missense variants were performed using PolyPhen,19 PolyPhen-2,19 MuPro,20 SIFT21 and AlignGVGD.22 23 Measurements of the evolutionary conservation of the nucleotide were performed by GERP (from the original SeattleSeq annotation) and PhyloP. The Grantham matrix score was used to measure how different the two amino acids (wildtype vs mutant) are to each other.24 Candidate genes with fetal nervous system expression (see the URL under Web resources) were verified by PCR/Sanger sequencing (data not shown).
Tissue expression
Clontech Multiple Tissue cDNA (Clontech, Mountain View, California, USA) panels from human fetal (Cat. #: 636747) and adult (Cat. #: 636742) tissues were used for expression analysis. Per Clontech certificate of analysis, the cDNA panels were generated from poly A+ RNA from the various tissues with assurance of original DNA contamination of <0.01% by pico green staining. Five μl (∼5 ng) of cDNA was mixed with 10 μl each (5 μM) of forward and reverse primers with 25 μl of 2× PCR Master mix (Cat. #: M7502; Promega, Madison, Wisconsin, USA). RAB40AL specific primers 5′ GCCTGCAGGACGGCACGGCC 3′ and 5′ TCCCTCTTCCAGGTGTAACCATT 3′ were used along with human β-actin gene (ACTB) primers 5′ ATATCGCCGCGCTCGTCGTC 3′ and 5′ AGTGGTACGGCCAGAGGCGT 3′. PCR conditions were 95°C for 2 min, followed by 30 cycles of 95°C for 30 s, 60°C for 1 min, 72°C for 1 min, and 72°C for 5 min. The PCR product was analysed by agarose gel electrophoresis.
GFP expression vector construction
pmCIT–C1 and pmCIT–N1 vectors (derivatives of Clontech pEGFP–C1 and –N1 vectors)25 were used for generating expression constructs with RAB40AL tagged with green fluorescent protein (GFP). Standard molecular biology techniques were employed for engineering the constructs. The single exon RAB40AL was PCR amplified from human male control or patient DNA using primers 5′ AGATCTATGAGCGCCCCGGGCAGCCCCGAC 3′ and 5′ GTCGACTTAAGAAATTTTGCAGCTGTTTCT 3′, or 5′ CTCGAGATGAGCGCCCCGGGCAGCCCCGAC 3′ and 5′ AAGCTTAGAAATTTTGCAGCTGTTTCT 3′ (restriction sites underlined). The first primer set engineers BglII and SalI restriction sites at the 5′ and 3′ ends of the PCR product, respectively. These sites allow for inframe cloning of GFP and RAB40AL wild type or p.D59G variant coding sequence in pmCIT–C1 to generate N-terminal fusions. The second set of primers engineers XhoI and HindIII restriction sites at the 5′ and 3′ ends of the PCR products. These PCR products were cloned into pmCIT–N1 to generate C-terminal inframe fusions of GFP to RAB40AL wild type or p.D59G variant. We used GFP–RAB40AL and GFP–RAB40AL–p.D59G to denote the N-terminal tagged constructs, and RAB40AL–GFP and RAB40AL–p.D59G–GFP to denote C-terminal tagged constructs. Vector constructs were confirmed by Sanger sequencing for proper engineering and sequence accuracy.
Transfection and immunocytochemistry
COS7 cells were grown in DMEM with 10% FBS in a 12-well plate on round coverslips. At ∼90% confluence, transfection was performed with lipofectamine 2000 (Invitrogen, Carlsbad, California, USA). Specifically, 2 μg of vector DNA in water was diluted in 100 μl of Opti-MEM reduced serum media (GIBCO, Carlsbad, California, USA) for 5 min at room temperature. In addition, 5 μl of lipofectamine reagent was mixed with 100 μl of Opti-MEM reduced serum media and incubated for 5 min at room temperature. Both solutions were then combined and incubated for 20 min at room temperature. Subsequently, an additional 300 μl of Opti-MEM reduced serum media was added to the mix and the total volume then added to cells and incubated at 37°C for 4 h. Subsequently, 500 μl of DMEM with 10% FBS was added to the coverslips and cells incubated at 37°C for 48 h. Cells were then fixed with ice-cold methanol for 5 min, air dried, washed with PBS followed by two washes with PBS/Tween 20 (0.1%) 5 min each, and then treated with 1:500 dilution of mouse anti-human mitochondrial COXIV antibody (Cat. #: mAbcam33985; Abcam, Cambridge, Massachusetts, USA) and incubated overnight at 4°C. Following two more washes with PBS/Tween-20, Alexa Fluor 555 F(ab')2 fragment of goat antimouse IgG (Cat. #: A21425; Invitrogen) diluted 1:500 was added and incubated for 1 h at room temperature. Following two additional washes, cells were stained with DAPI (1:500 dilution in PBS), mounted and images captured on a Leica DM5000B microscope (W. Nuhsbaum Inc., McHenry, Illinois, USA). Transfection efficiencies were measured by determining the percentage of GFP+ cells among total DAPI+ cells transfected with vector only, WT construct or MUT construct. Nuclear clustering percentages were measured by determining the percentage of cells with fusion protein within the nucleus, nucleolus and/or perinuclear regions among all GFP+ cells.
Cell lysate preparation, PAGE and western analysis
Confluent COS7 cells in 12-well tissue culture plates were washed twice with ice-cold PBS and treated with 500 μl of RIPA buffer (Cat. #: R0278; Sigma, St. Louis, Missouri, USA) with 1% Protease Inhibitor Cocktail BD BaculoGold (Cat. #: 554779; BD Biosciences, San Diego, California, USA). After thoroughly mixing, the cell suspension was incubated on ice for 20 min with additional vortexing for complete lysis. The suspension was sonicated for 10 min and then centrifuged at 13 000 rpm for 5 min. The supernatant was collected and quantified using Bio-Rad Protein Assay kit (Cat. #: 500-0006; Bio-Rad, Hercules, California, USA). Approximately 30 μg of protein was added to 5 μl of NuPAGE sample buffer (Cat. #: NP0007; Invitrogen) along with 2 μl of NuPAGE reducing agent (Cat. #: NP0004; Invitrogen). The sample mix was heated for 10 min at 80°C. Precast Invitrogen NuPage 4–12% Bis-Tris gels (Cat. #: NP0322BOX; Invitrogen) were used for polyacrylamide gel electrophoresis (PAGE). NuPAGE antioxidant (0.25%) (NP0005; Invitrogen) was added to 200 ml of 1X MOPS (Cat. #: NP0001; Invitrogen) running buffer and poured in the centre of the apparatus. Untreated 1X MOPS was used in the remaining chamber. The gel was run at 200 V for 1.5 h and proteins transferred onto Immobilon PVDF transfer membranes (IPSN07852; Millipore, Billerica, Massachusetts, USA) using NuPage Transfer buffer (Cat. #: NP0006-1; Invitrogen). Membranes were blocked overnight at 4°C with 5% BSA prepared in tris-buffered saline. Subsequently, the membranes were incubated for 2 h at room temperature with the primary mouse anti-GFP monoclonal antibody (Cat. #: 632380; Clontech) (or primary mouse anti-glyceraldehyde-3-phosphate dehydrogenase monoclonal antibody for detecting control house-keeping protein; Cat. #: MAB374, Millipore, Temecula, California, USA) followed by five washes (5 min each) with tris-buffered saline–0.5% Tween20. The membrane was then incubated with secondary anti-mouse horseradish peroxidase-conjugated antibody (Cat. #: 1858415; Pierce/Thermo Scientific, Rockford, Illinois, USA) for 1 h at room temperature. Washes were repeated and protein was detected using the chemiluminescence system (Cat. #: 34087; Thermo Scientific).
Statistical analysis of data
The SPSS Statistics program V.19 was used for statistical analysis of data. A general linear model was performed to examine: (1) transfection efficiencies (number of GFP+ cells/number of DAPI+ cells) among the three vectors and (2) the number of transfected cells with clustering of GFP fluorescence signal within the nucleus, nucleolus and/or perinuclear regions for the three vectors. Post hoc comparisons were performed to determine differences among the three groups with a Bonferroni correction.
Results
RAB40AL is mutated in MPS
WGS, WES and X chromosome specific exome sequencing were performed to analyse DNA from two related male subjects affected with MPS (WGS, WES and X exome sequencing were performed for individual III-5, and X exome sequencing only was performed for individual IV-1; see table 1 and figure 1A). The average read depths per target region were 4.8X and 10.9X for WGS and WES, respectively, and 86.9X (for III-5) and 17.9X (for IV-1) for X-specific exome sequencing. Combining all three massively parallel sequencing strategies provided complete sequence coverage for 96.4% (3496/3625) of exons with ≥4× depth of coverage. Sixty-one of the remaining 129 exons were not covered by the three different sequencing strategies, yet were expressed in the fetal nervous system; the other 68 exons were not expressed in the human fetal nervous system and were excluded from further analysis. Forty-four of the remaining 61 exons were Sanger sequenced and revealed no potentially damaging or pathogenic variants (Supplemental table 1). Seventeen exons proved difficult to sequence after multiple amplification attempts with different PCR primer pairs, presumably due to their high repetitive sequence nature and/or higher GC content.
Summary of whole genome, whole exome and X chromosome specific exome sequencing for two affected related individuals
Massively parallel sequencing analysis identified 15 candidate variants in 10 genes common to III-5 and IV-1 (table 1). Two genes, RAB40AL and ARHGEF9, contained missense mutations. Application of multiple protein function prediction algorithms showed the RAB40AL p.D59G variant (102,079,078-102,079,079AC→GA, hg18) to be highly damaging (table 1 and Supplemental table 2), while the ARHGEF9 p.D306E variant (62,810,649G→C, hg18) was not considered damaging. RAB40AL was covered 100% with optimum coverage noted using the X-specific exome sequencing strategy (Supplemental figure 1.
The RAB40AL p.D59G variant segregated with the phenotype in MPS family members (figure 1A), and all obligate and non-obligate female subjects with X chromosome skewing were carriers of the mutation.13 The AC→GA change in hemizygous male subjects and heterozygous female subjects and the resulting D59G amino acid substitution are shown in figure 1B. Asp59 (D59) is highly conserved from humans to mosquitoes (figure 1C). There were no p.D59G variants detected after: (a) direct sequencing of 297 neurologically normal control male subjects (250 of 297 were Caucasian) obtained from the Greenwood Genetic Center, National Institute of Neurological Disease and Coriell Institute for Medical Research and (b) analysis of the most recent 1000 Genomes Project dataset or the National Heart, Lung, and Blood Institute Exome Sequencing Project (∼3000 individuals of unknown gender; 10/2011 accessed). Based on published analytical approaches,26 this number of controls provides >95% power to exclude RAB40AL p.D59G as a normal sequence variant with 0.1% frequency.
RAB40AL appears to be a primate innovation
In mammals, X chromosome genes maintain complete synteny because of the constraints imposed by X chromosome inactivation; however, some non-pseudoautosomal X chromosome genes violate this rule.27 Detailed analysis of the corresponding syntenic region identified RAB40AL orthologues in primates such as rhesus monkeys and chimpanzees, but not in dog, rat, mouse, Xenopus or zebrafish (data not shown). Nucleotide- (using both 5′- and 3′-UTR regions as well as the complete gene) and protein-BLAST analyses of RAB40AL and RAB40AL, respectively, identified mouse Rab40b as the gene product with the highest identity (83%) and similarity (91%) to human RAB40AL; however, the orthologue of Rab40b on mouse chromosome 11 is RAB40B on human chromosome 17 (figure 2; confirmed by synteny analysis). Human RAB40A shows 99% protein identity and similarity to RAB40AL; the RAB40A gene is ∼560 kb telomeric to RAB40AL on the X chromosome and is flanked by repetitive sequences. The phylogenetic relationship of the RAB40 family of proteins is shown in figure 2. The apparent restriction of RAB40AL to primates suggests a recent evolutionary divergence and is consistent with the hypothesised functional role of RAB40AL in neurodevelopment.

Phylogenetic relationship of RAB40 family of proteins. Dendogram generated from protein sequences aligned using CLUSTAL W V.2.1 is shown. Synteny analyses of RAB40AL and RAB40A using UCSC Genome Browser did not identify mouse, Xenopus or zebrafish orthologues of human RAB40AL. The X-linked RAB40AL and RAB40A are unique to primates (red rectangle). Un, unknown.
RAB40AL is expressed in fetal and adult human tissues including brain and kidney
Because individuals with MPS exhibit developmental delay and cognitive impairment, we hypothesised that RAB40AL should be expressed in both fetal and adult brain tissues. Analysis of RNA from fetal and adult human tissues using RT-PCR showed that RAB40AL is expressed in MPS-related tissues, including brain and kidney (figure 3), consistent with reported MPS phenotypes.12 Based on this semiquantitative PCR-based expression analysis, RAB40AL expression levels in the fetal lung, heart, liver and skeletal muscle appeared lower than in adult tissues (figure 3), but this needs to be confirmed by quantitative methods. Others have also shown, by northern analysis, that RAB40AL is expressed in adult brain, heart, liver, skeletal muscle and kidney.11

RAB40AL expression in relevant fetal and adult human tissues. RT-PCR using Clontech multiple tissue cDNA panels from human fetal and adult tissues shows high RAB40AL expression in fetal and adult brain and kidney tissues, with lower levels of expression in fetal lung, heart, liver and skeletal muscle. Single bands corresponding to 812 bp and 437 bp for RAB40AL and β–actin PCR products, respectively, were observed as expected. Bands were also confirmed by Sanger sequencing. A, adult; F, fetal.
The D59G missense change alters RAB40AL abundance in COS7 cells
Based on the location of D59 within the GTPase domain of RAB40AL, specifically within a conserved loop region located between two presumptive β-strands, we predicted that the D59G missense mutation might render it unstable, reducing function. The C-terminal HVR of RAB40AL contains sequence motifs that mediate attachment of lipid moieties predicted to anchor it to the phospholipid bilayer. Consequently, an unconstrained C-terminus may be critical for normal RAB40AL biological function. To test this, we constructed N-terminal and C-terminal GFP-tagged fusions of RAB40AL and RAB40AL–p.D59G and examined the impact of p.D59G by transient transfection and western analysis. Results of western analysis using anti-GFP monoclonal antibody of whole cell lysates from cells transiently transfected with these expression vectors are shown in figure 4. Both GFP (control vector alone) and WT GFP–RAB40AL were easily detected. In contrast, MUT GFP–RAB40AL–p.D59G fusion protein was significantly reduced compared with WT, despite similar transfection efficiencies (see table 2). We hypothesise that the reduced abundance of GFP–RAB40AL–p.D59G is due to instability and degradation of the fusion protein.

Reduced abundance of green fluorescent protein (GFP)–RAB40AL–p.D59G fusion protein in COS7 cells. Western analysis of whole cell lysates from cells transiently transfected with GFP–RAB40AL vector constructs (V, vector only; WT, GFP–RAB40AL; or MUT, GFP–RAB40AL p.D59G) using anti-GFP (or anti-GAPDH) monoclonal antibody (mAb). Results show reduced abundance of GFP–RAB40AL–p.D59G fusion protein, with essentially unchanged levels of the house-keeping GAPDH protein. Transfection efficiencies were not significantly different among the three vector constructs (see table 2). COS, untransfected COS7 cell lysate.
Quantitative analysis of transfection experiments
We observed low abundance bands from both RAB40AL–GFP and RAB40AL–p.D59G–GFP constructs, despite similar transfection efficiencies (data not shown). The low abundance of RAB40AL–GFP fusion protein suggests that constraining the RAB40AL C-terminus with a bulky (∼27 kDa GFP) protein disrupts RAB40AL function and renders it unstable. However, further analyses are needed to directly measure RAB40AL synthesis and degradation kinetics.
The D59G missense change disrupts RAB40AL cytoplasmic localisation
Previous studies have shown that RAB40AL localises to the mitochondria in COS7 cells.11 In addition, analysis of the C-terminal 49 amino acid HVR of RAB40AL by MitoPred, a mitochondrial prediction algorithm, predicted mitochondrial localisation with 99% confidence (data not shown). This was not the case for Xenopus Rab40c or human RAB40C, where the former is known to be localised to the Golgi.28 Consequently, we examined whether the p.D59G variant disrupts this mitochondrial localisation (figure 5 and Supplemental figure 2). As expected, GFP–RAB40AL localised to the mitochondria but was also found diffusely throughout the cytoplasm (figure 5, panel H). This cytoplasmic localisation was disrupted in GFP–RAB40AL–p.D59G constructs and the fusion protein appeared to be accumulated or clustered within the nucleus, nucleolus and/or perinuclear region (figure 5, panels I and L). One-way ANOVA revealed that cells transfected with V, WT and MUT constructs were significantly different on ‘nuclear clustering’ efficiency (F2,26 = 41.074, p<0.001). Pairwise post hoc comparison indicated that cells transfected with the MUT construct (64.0±16.9; mean±SD) were significantly different than those transfected with WT (30.1±16.2; mean±SD) for per cent nuclear clustering (p<0.001) (see also table 2). Interestingly, the C-terminal GFP-tagged fusions (RAB40AL–GFP and RAB40AL–p.D59G–GFP) exhibited staining patterns similar to the N-terminal tagged mutant fusion construct (GFP–RAB40AL–p.D59G) (compare panels I and L in Supplemental figure 2 with panel L in figure 5). Thus, the D59G missense change appears to disrupt the normal localisation of RAB40AL, perhaps via mechanism that also render it prone to degradation (figure 4). These data also suggest that the RAB40AL C-terminus is critical for protein stability and intracellular protein localisation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
p.D59G disrupts normal intracellular protein localisation. Cells transiently transfected with N-terminal green fluorescent protein (GFP)-tagged vector constructs (vector control, A-D; RAB40AL, E-H; and RAB40AL p.D59G, I-L) and stained with anti-mitochondrial COXIV antibody (Mito Ab) and DAPI are shown. GFP–RAB40AL localises to the mitochondria and throughout the cytoplasm (panel H), while this localisation is disrupted with GFP–RAB40AL–p.D59G (panel L). GFP–RAB40AL–p.D59G appears to be accumulated or clustered within the nucleus, nucleolus and/or perinuclear region (panels I and L). Representative transfected cells are shown (arrows).
Discussion
We have identified a germline missense RAB40AL mutation (p.D59G) in individuals affected with MPS. This variant is predicted to be damaging using seven different DNA/protein functional inference algorithms, is absent in ∼3300 normal controls, segregates with the phenotype in the family and is located within a highly conserved GTPase region of the protein. RAB40AL is expressed in fetal and adult brain tissues, consistent with the cognitive impairments present in MPS individuals. Functional analysis of the p.D59G variant by transfection studies and western blotting showed that the variant renders the protein unstable and disrupts its normal cytoplasmic localisation. An unconstrained C-terminus appears necessary for normal RAB40AL localisation, based on transient transfections of N-terminal and C-terminal tagged wild type and mutant fusion constructs. We conclude that disruption of RAB40AL function by the p.D59G variant leads to MPS.
XLID, X- linked hearing loss and X-inactivation
MPS is one among approximately 200 X- linked intellectual disability (XLID) disorders.29 The prevalence of XLID is approximately 1:1000 in male subjects, and XLID accounts for ∼16% of male subjects with intellectual disability.29 30 Among the 1606 genes on the human X chromosome (Build 37.1), at least 87 have been identified as causative for XLID.29 31 32 Several genes encoding GTP-binding proteins are reported to play critical roles in the development and function of the CNS, including several that participate in intracellular signalling or vesicular trafficking.33–37 Data reported here support the hypothesis that RAB40AL is critical for normal development and function of CNS and RAB40AL can now be added to the list of syndromic XLID causative genes.
In addition to cognitive impairments, individuals with MPS have bilateral congenital sensorineural hearing loss.12 X- linked forms of hearing loss account for ∼5% of all congenital hearing loss, and at least eight different causative genes have been described (DFN1/TIMM8A, DFN2/PRPS1, DFN3/POU3F4, DFN6/SMPX, DFN7/GSTP1, NDP, FLNA and MED12).12 29 Mutations in TIMM8A, which encodes a mitochondrial intermembrane protein that interacts with a GTPase to regulate programmed cell death, causes Mohr–Tranebjaerg/deafness–dystonia syndrome.38 Interestingly, several autosomal-linked small GTPase proteins, including the Rho GTPases, also contribute to hearing phenotypes.39 40 To our knowledge, ours is the first study to show that mutation of RAB40AL is associated with hearing loss.
X-inactivation equalises the dosage between male and female subjects for most X chromosome genes, and this process is normally random in cells. In the presence of certain gene mutations and genomic alterations, the X chromosome bearing the altered gene or genomic region in female subjects is preferentially inactivated. Exceptions to this rule include cases where an X chromosome is translocated onto an autosome.41 In MPS, four obligate and four non-obligate female heterozygous D59G mutation carriers show skewed X-inactivation and are clinically unaffected.12 13 Interestingly, the unaffected mother of the male individual described by Saito-Ohara et al (2002)11 was a carrier of the same inversion (46,X, inv(X)(p21.2q22.2)) but her X-inactivation status was not reported. Consequently, X chromosome inactivation skewing correlates with heterozygosity for the p.D59G variant and is another useful indicator of carrier status in MPS.
The RAB40AL p.D59G variant
Insights into the potential effect of D59G on RAB40AL function can be gained by examining the characteristics of the D47 aspartate residue (equivalent to RAB40AL D59) relevant for Rac other Ras-like proteins. D47 is predicted to be highly relevant for Rab function.42 In addition to the known canonical function of GTPases of cycling between two states (ie, GDP vs GTP bound), phosphorylation of specific C-terminal serine residues in Ras proteins is thought to act as an allosteric discriminator of conformational/dynamic states. Phosphorylation influences effector protein interactions with the GTPase protein at the switch loops/effector domain.43 D47 in HRAS and RAP1B (another Ras-like protein) is located between the conserved β-2 and β-3 strands, and appears to be involved in facilitating this switch in conformational/dynamic states (from inactive to active) following S179 phosphorylation in the HVR domain.43 Removal of D47 or E49 in HRAS negatively influences Ras signalling and orientation in the membrane.44 The p.D59G variant may disrupt protein folding, nucleotide-binding (consequently, GTPase activity) or localisation rendering the protein non-functional and prone to degradation.
The Rab40 family
Current knowledge of the characteristics and roles of the various Rab40 proteins is limited. To date, the best characterised of the Rab40 proteins are the Rab40c family members. In Xenopus, Rab40c is required for normal gastrulation and embryogenesis.28 Rab40c is localised at the Golgi apparatus and interacts with ElonginB/C and Cullin5 to form a ubiquitin ligase complex that selectively polyubiquitinates Rap2 and regulates non-canonical Wnt signalling.28 In rat, Rab40c is expressed in oligodendrocytes and is localised to the recycling endosomal compartment.45 Human RAB40C is regulated directly by a cancer-related miRNA, let-7a, and mediates the biological effects of let-7a in gastric tumorigenesis.46 Thus, Rab40c proteins have diverse functions in embryogenesis, ubiquitination and cancer.
In contrast to Rab40c, Rab40a and Rab40b function and intracellular localisation remain to be determined. Rab40b is expressed in the zebrafish brain (zebrafish database), suggesting a role in neurodevelopment. We found that mouse Rab40b is expressed in E14.5 embryonic and adult mouse brain, inner ear and heart tissues (data not shown). It remains to be determined, however, whether humans with mutated RAB40B also exhibit cognitive impairment and hearing loss, similar to individuals with MPS.
Unlike RAB40C and RAB40B, RAB40A and RAB40AL appear to be primate innovations (figure 3). A key approach for identifying orthologues of human genes in other species is synteny analysis, that is, exploiting the gene order conservation between species.47 48 This approach identified orthologues of RAB40AL in primates only. Interestingly, primate specific genes display distinctive features including high tissue specificity, rapid evolution and short peptide size.48 Some primate-specific genes are thought to have been formed by gene duplication.48 and transposable elements also appear to play an important role.48 49 Whether gene duplication or transposon-mediated events gave rise to RAB40AL in primates is not clear. There are at least several hundred genes that differ between human and mouse, most as a result of gene duplications or deletions.50 51 The extent to which RAB40AL function overlaps with the other Rab40 members or is unique to primates is currently unknown. Study of the paralogue of RAB40AL (Rab40b) or introduction of human RAB40AL mutations into the mouse may provide insights into the roles of RAB40B, RAB40A and RAB40AL in brain, inner ear and other organ development.
Summary
Using comprehensive variant discovery by high-throughput sequencing, we have identified RAB40AL p.D59G in the multi-organ system neurodevelopmental disorder MPS. The specific signalling pathway for RAB40AL and its particular mechanism and function in the mitochondria remain to be elucidated. Identification of RAB40AL mutations allows for detailed investigations into its molecular mechanism and functions in MPS, and should help improve diagnosis and treatment of individuals with related phenotypes.
Web resources
The URLs for data presented are as follows:
AlignGVGD; http://agvgd.iarc.fr/agvgd_input.php
CLUSTAL W version 2.1; http://www.genome.jp/tools/clustalw/
Human fetal nervous system expression database; http://bgee.unil.ch/bgee/
MitoPred, http://bioapps.rit.albany.edu/MITOPRED/
NHLBI Exome Sequencing Project (Exome Variant Server); http://snp.gs.washington.edu/EVS/
PhyloP; http://genome.ucsc.edu/
PICARD; http://picard.sourceforge.net/
PolyPhen; http://genetics.bwh.harvard.edu/pph/
PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/
SeattleSeq; http://gvs.gs.washington.edu/SeattleSeqAnnotation/index.jsp
SIFT; http://sift.jcvi.org/
UCSC Genome Browser; http://genome.ucsc.edu
Zebrafish database; http://zfin.org
Acknowledgments
We thank Drs Susan L Dagenais and Robert H Lyons from the UM DNA Sequencing Core for help with sequencing, members of the Martin laboratory for their technical help, Dr Christopher Vlangos for providing the COS7 cells, Jillian Lee Wiggins for help with the statistical analyses, and Dr Rick Neubig for critically reading this manuscript. pmCIT–C1 and pmCIT–N1 vectors were generously provided by Dr Lois Weisman at the University of Michigan.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
Funding VMS was supported by the Genome Sciences Training Grant. This work was supported by NIH T32 DC00011 (JAM), NIH K12 HD028820 (JKB), an award from the Elizabeth E Kennedy (Children's Research) Fund through the Department of Pediatrics (JKB), an award from the University of Michigan Center for Genetics in Health and Medicine (DMM and JZL), an award from the Michigan Institute for Clinical and Health Research (DMM), and NIH R01 grants NS054784 and DC009410 (DMM).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.