Article Text

Abstract
Background Fanconi anaemia (FA) is a group of disorders characterised by progressive bone marrow failure and a characteristic but variable craniofacial and skeletal involvement. Recessive mutations in any of 15 genes linked to FA lead to the pathognomonic increased susceptibility to double-strand DNA breaks.
Methods Autozygome and exome analysis of a patient with classic FA phenotype
Results The authors identified a novel truncating mutation in XRCC2. Consistent with the proposed causal link to FA, this gene is an essential non-redundant component of the RAD51 family of homologous repair proteins and its deficiency in a murine model has been shown to lead to a highly similar phenotype to that of this patient both at the cellular and organismal level.
Conclusion This study implicates XRCC2 in the pathogenesis of FA and calls for further investigation of the potential contribution of XRCC2 mutations to the overall mutational load of FA.
- Autozygome
- double-strand DNA breaks
- Fanconi anaemia
- genetics
- RAD51
Statistics from Altmetric.com
Fanconi anaemia (FA) is a group of disorders characterised by progressive bone marrow failure and a characteristic but variable craniofacial and skeletal involvement. Consistent with the pathognomonic finding of double-strand DNA breaks in the chromosomes of these patients when cultured with DNA interstrand crosslinking agents, all FA genes (15 to date) are known to play a role in dsDNA breaks repair.1 2 Although recessive mutations in these genes account for the majority of FA cases, the genetic heterogeneity of this disorder has not yet been fully captured.
In the highly consanguineous population of Saudi Arabia, we have shown marked allelic as well as genetic heterogeneity for a large number of Mendelian disorders, and how autozygome analysis can be a powerful tool to identify the underlying genetic defect.3 In this communication, we describe a Saudi child with a novel FA group in whom the combined use of autozygome and exome analysis led to the identification of a XRCC2 mutation as the likely cause of his phenotype.
Subjects and methods
Human subject
The patient had standard haematological and clinical genetics evaluation. He and his parents were enrolled in an Institutional Review Board-approved research protocol with written informed consent. This was followed by blood extraction in EDTA (for DNA extraction) and sodium-heparin (for Epstein–Barr virus-transformed lymphoblast cell line) tubes.
Autozygome analysis
This was performed as described before.4 5 Briefly, DNA was extracted from the patient and his parents and processed for genotyping using the Axiom platform following the manufacturer's protocol (Affymetrix, Santa Clara, CA, USA). Resulting genotypes were analysed using autoSNPa (http://dna.leeds.ac.uk/autosnpa/) to determine the overall pattern of autozygosity as judged by the runs of homozygosity (ROH) per individual (autozygome).6 We have only considered runs of homozygosity that are 2 Mb or more and contained at least 175 single-nucleotide polymorphisms to increase the likelihood that these ROH represent autozygous (identical by descent) rather than identity by state.
Exome analysis
Exome capture was performed using the TruSeq Exome Enrichment kit (Illumina, San Diego, CA, USA) following the manufacturer's protocol. Samples were prepared as an Illumina sequencing library, and in the second step the sequencing libraries were enriched for the desired target using the Illumina exome enrichment protocol. The captured libraries were sequenced using the Illumina HiSeq 2000 sequencer. The reads are mapped against UCSC hg19 (http://genome.ucsc.edu/) by Burrows–Wheeler Aligner (http://bio-bwa.sourceforge.net/). The single-nucleotide polymorphisms and indels are detected by SAMTOOLS (http://samtools.sourceforge.net/).
Results
Case report
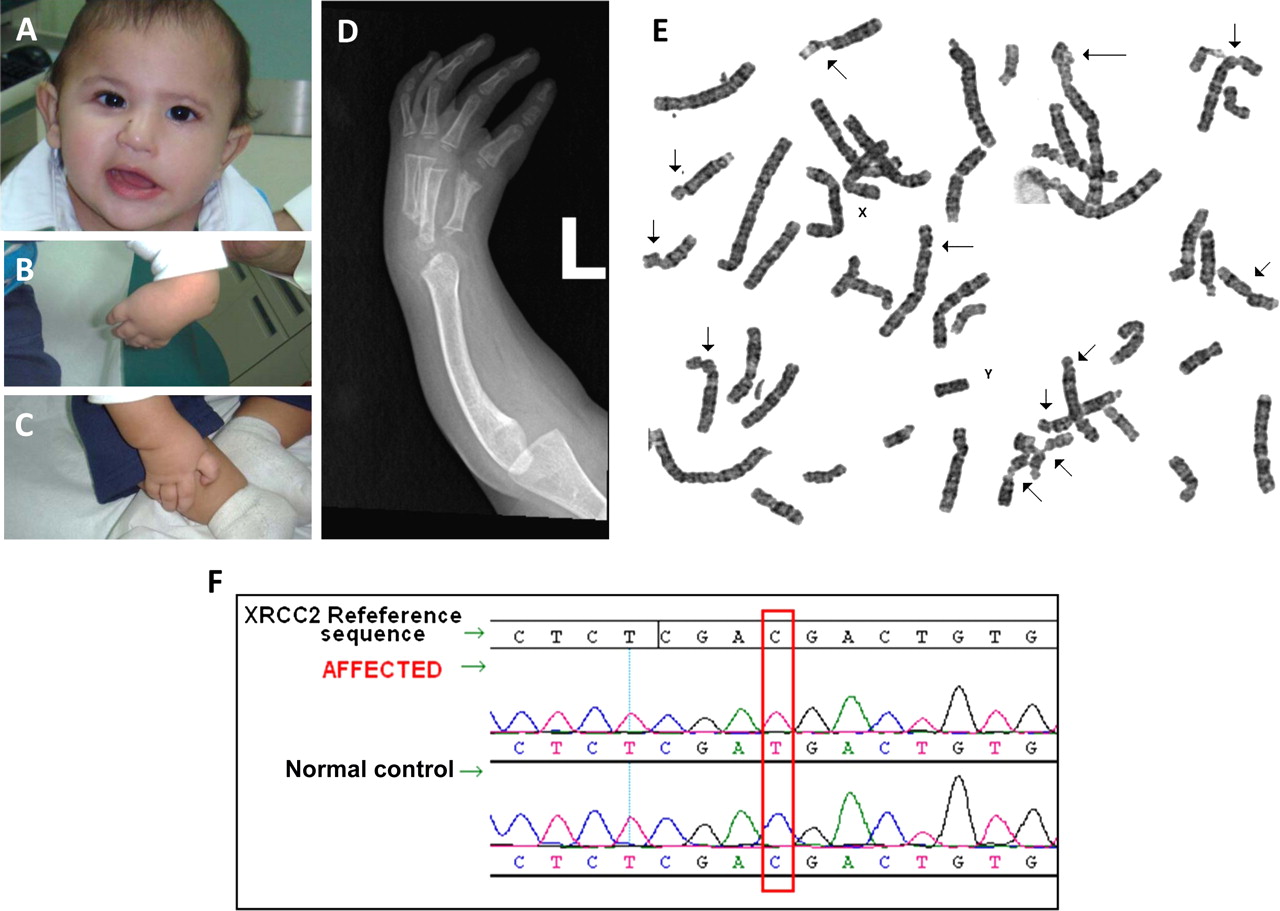
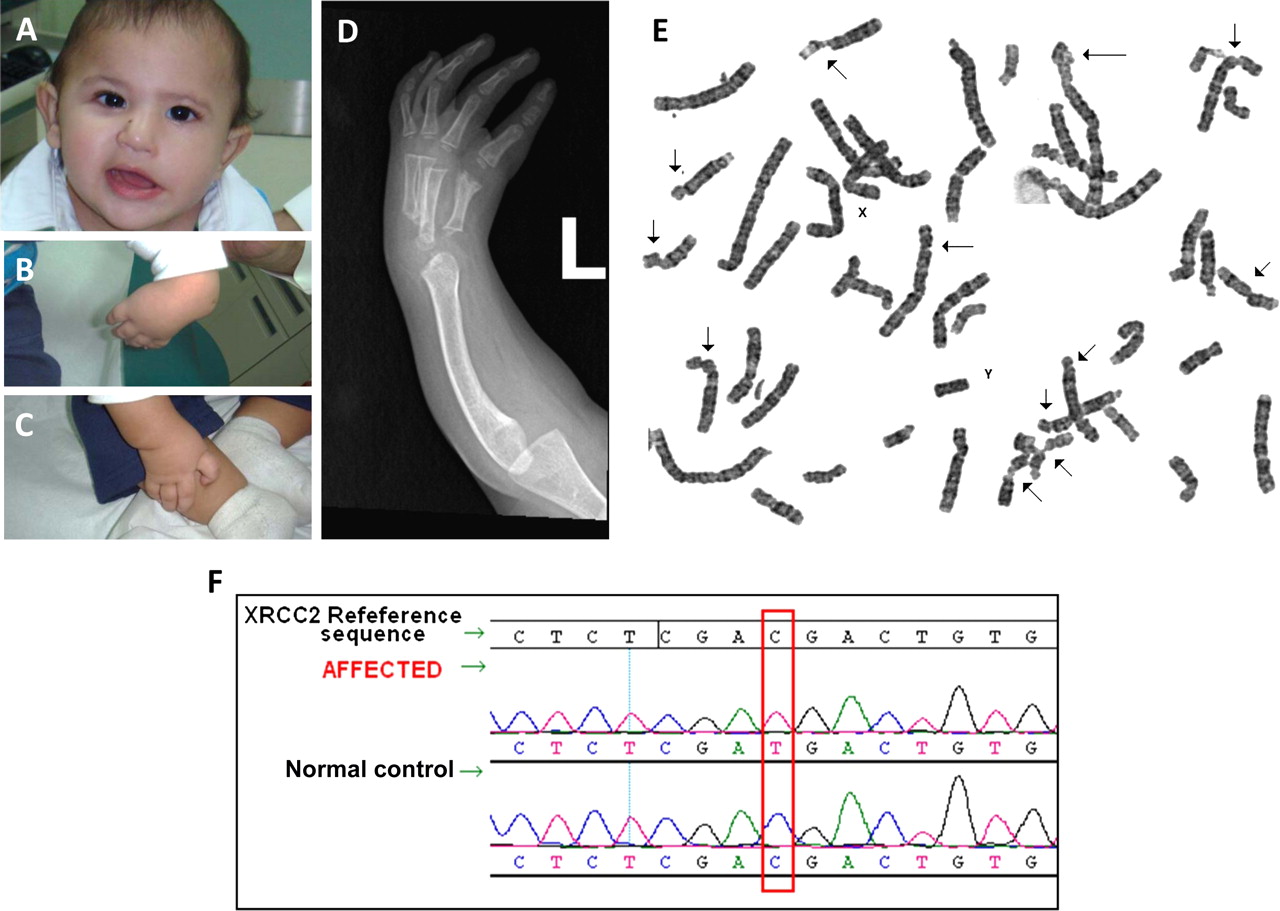
Index is a 2.5-year-old Saudi child who was born to young healthy first cousin Saudi parents. The pregnancy was unremarkable and he was born at term. Length and head circumference were abnormally low at birth (length 41 cm, weight 2.6 kg and head circumference 30.5 cm). His physical examination revealed left facial nerve palsy, microcephaly and bilaterally absent thumbs. Investigations in the nursery revealed ectopic left kidney, haemodynamically stable patent ductus arteriosus, normal blood indices and blood karyotype. Skeletal survey revealed complete absence of the first metacarpal and scaphoid bones bilaterally, absent left radius and hypoplastic right radius. On follow-up visits, he displayed severe growth deficiency and microcephaly. In addition, the diepoxybutane (DEB) test showed marked increase in the frequency of dsDNA breaks (figure 1).
{kind=link}
(A) Facial photograph showing facial palsy. (B) and (C) Hand photographs documenting lack of thumbs. (D) x-Ray of the left hand showing agenesis of the radius. (E) Chromosomal breakage study with diepoxybutane (DEB) showing the numerous sites of dsDNA breaks (arrows) typical of Fanconi anaemia. (F) Sequence chromatogram showing a novel homozygous XRCC2 mutation in the patient.
Exome and autozygome analysis
In view of the genetic heterogeneity of FA, we took advantage of the consanguineous nature of the parents to pursue autozygome analysis in order to identify the most likely candidate.7 Supplementary figure S1 and table S1 (available online only) summarise the overall ROH pattern of the patient. FANCL was the only autosomal recessive FA gene to reside within ROH but we failed to identify any mutation in that gene at the genomic or complementary DNA level. This led us to hypothesise that this child's FA is caused by a recessive mutation of a novel FA gene. Therefore, we set out to perform whole exome sequencing followed by autozygome filtering.4 5 Supplementary table S2 (available online only) summarises the raw data of our exome sequencing. Our filtration strategy (summarised in supplementary figure S2, available online only) allowed us to narrow the search to three coding variants: MTBP (NM_022045:c.A397G; p.(N133D)), RGS3 (NM_144488: c.C2882A; p.(T961N) and XRCC2 (NM_005431.1:c.643C>T; p.(Arg215*)). The first two missense variants were deemed unlikely because they were detected in 1/270 and 3/192 control Saudi samples, respectively. The truncating nonsense mutation in XRCC2 was therefore the only likely variant left based on our filtration strategy.
Discussion
XRCC2 is one of five RAD51 paralogs that act non-redundantly in the same pathway of homologous recombination repair (HR) of dsDNA breaks.8 It has been shown that XRCC2 enhances the binding of RAD51 to sites of dsDNA breaks, a key step in HR, and truncation mapping is consistent with the hypothesis that our patient's truncation is pathogenic as it removes an important C-terminus, lack of which fully abolishes XRCC2 activity.9 XRCC2 deficiency has been repeatedly shown at the cellular level to result in impaired HR and increased susceptibility to dsDNA breaks, consistent with our patient's chromosomal phenotype.8 Importantly, Xrcc2 knockout mice have embryonic lethality and abnormal patterning leading to multiple congenital malformations (including microcephaly and skeletal anomalies) that resemble our patient's phenotype.10 Finally, the severe proliferation disadvantage of fibroblasts derived from these mice is also consistent with our observation on skin-derived fibroblasts from our patient.11
Our results, therefore, strongly implicate XRCC2 in the pathogenesis of FA and call for further investigation of the potential contribution of XRCC2 mutations to the overall mutational load of FA. This case shows the power of next generation sequencing when combined with autoyzygome analysis in identifying novel genetic causes of Mendelian disorders even on simplex cases.
Acknowledgments
The authors would like to thank the family for their enthusiastic participation. They also wish to thank the genotyping and sequencing core facilities at KFSHRC for their technical help.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
Funding This study was funded in part by KACST grant 09-MED941-20 (FSA), and a Dubai-Harvard Foundation for Medical Research Collaborative Grant (FSA).
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by the King Faisal Specialist Hospital and Research Center Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.