Article Text

Abstract
Dyskeratosis congenita (DC) is a premature ageing syndrome characterised by short telomeres. An X-linked form of DC is caused by mutations in DKC1 which encodes dyskerin, a telomerase component that is essential for telomerase RNA stability. However, mutations in DKC1 are identifiable in only half of X-linked DC families. A four generation family with pulmonary fibrosis and features of DC was identified. Affected males showed the classic mucocutaneous features of DC and died prematurely from pulmonary fibrosis. Although there were no coding sequence or splicing variants, genome wide linkage analysis of 16 individuals across four generations identified significant linkage at the DKC1 locus, and was accompanied by reduced dyskerin protein levels in affected males. Decreased dyskerin levels were associated with compromised telomerase RNA levels and very short telomeres. These data identify decreased dyskerin levels as a novel mechanism of DC, and indicate that intact dyskerin levels, in the absence of coding mutations, are critical for telomerase RNA stability and for in vivo telomere maintenance.
- Telomerase
- dyskerin
- dyskeratosis congenita
- pulmonary fibrosis
- genetics
- haematology (incl blood transfusion)
- respiratory medicine
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Telomerase
- dyskerin
- dyskeratosis congenita
- pulmonary fibrosis
- genetics
- haematology (incl blood transfusion)
- respiratory medicine
Introduction
Dyskeratosis congenita (DC) is a rare inherited disorder defined by a triad of mucocutaneous features: skin hyperpigmentation, nail dystrophy, and oral leukoplakia. In DC families, bone marrow failure affects the majority of patients by the age of 20, and is the primary cause of mortality in 70% of cases.1 Pulmonary fibrosis, liver cirrhosis, and cancer account for the remaining causes.1 DC is a heritable, Mendelian disorder. Considering its classic dermatologic diagnostic criteria, the X-linked form of DC is most common, comprising half of all cases.2 Mutations in DKC1, which encodes the dyskerin protein, underlie inheritance in 50% of X-linked families,3 4 leaving the remaining cases genetically uncharacterised. Whether the remaining families have non-coding DKC1 mutations or variants in other X-linked genes is not known.
Dyskerin is one of four proteins which form a complex with RNAs that contain a box H/ACA motif.5 This class of non-coding RNAs includes the RNA component of telomerase, hTR (also referred to as TERC), and H/ACA small nucleolar RNAs (snoRNAs).5 6 Missense mutations in DKC1 lead to defects in hTR biogenesis and stability, and patients with DKC1 mutations can have as little as 20% of normal hTR levels.7 8 In addition to its role in hTR stability, dyskerin uses H/ACA snoRNAs to guide the site specific pseudouridylation of ribosomal RNAs.5 This dual function of dyskerin has raised the possibility that in addition to insufficient hTR, X-linked DC patients may have compromised snoRNA levels and defects in ribosomal RNA function.9–11 However, in cells isolated from individuals with DKC1 missense mutations, decreased snoRNA levels and pseudouridylation defects have not been readily identified.7 8 12 Whether snoRNA defects may be specific to a subset of missense mutations in dyskerin, and not others, is not known.
DC falls on the severe end of a spectrum of syndromes of telomere shortening.13 Mutations in the essential components of the enzyme telomerase, hTERT, the telomerase reverse transcriptase, and hTR, cause haploinsufficiency and autosomal dominant DC.14 15 In autosomal dominant DC due to mutations in hTERT and hTR, the clinical phenotype is heterogeneous and patients frequently present with bone marrow failure, pulmonary fibrosis or liver disease in the absence of the classic mucocutaneous features.15 16 In addition to DKC1, hTR, and hTERT, mutations in the telomere binding protein gene TINF2 account for a subset of young onset DC cases.17 18 We identified an X-linked DC pedigree that presented as familial pulmonary fibrosis. Although the DKC1 sequence was intact, genome wide linkage analysis implicated the DKC1 locus, and we detected significantly decreased hTR and dyskerin protein levels. Our data suggest that intact dyskerin levels, in the absence of coding mutations, are critical for hTR stability and for telomere maintenance in X-linked DC and pulmonary fibrosis.
Methods
Subjects
The family was identified as part of the Vanderbilt Familial Pulmonary Fibrosis Registry based on the confirmed diagnosis of idiopathic interstitial lung disease in two or more members.19 The study was approved by the local institutional review boards of Johns Hopkins and Vanderbilt Universities. Written informed consent was obtained from all subjects. Confirmation of the pulmonary fibrosis diagnosis was based on clinical assessment of the proband and review of the medical history and records of related individuals. The average length of telomeres was measured in primary lymphocytes by flow cytometry and fluorescence in situ hybridisation (FISH) (Repeat Diagnostics, North Vancouver, BC, Canada), as described.19 Lymphoblastoid cell lines were generated from peripheral blood lymphocytes as described.20 Control lymphoblastoid cells were derived from healthy male donors or from an unaffected male relative.
X-inactivation and FISH analysis
We performed X-inactivation analysis using the HUMARA-PCR assay.21 Briefly, genomic DNA was digested with a methylation specific enzyme (HpaII). PCR was performed at the polymorphic androgen receptor locus which contains (CAG)n repeats. After PCR amplification, we determined allele size on an ABI Genome Analyzer, and measured the ratio of methylated to unmethylated alleles. We performed metaphase FISH analysis using a probe mixture containing the Xq subtelomere (ToTelVysion Subtelomeric probe set, Vysis/Abbott Molecular, Des Plaines, Il, USA).
Sequencing
The 15 DKC1 exons, 3′UTR and promoter regions were amplified and sequenced using the listed primers (supplementary tables 1 and 2). Sequences were manually inspected (Sequencher v.4.9, Gene Codes, Ann Arbor, MI, USA), and variants were compared with entries in dbSNP build 130 (http://www.ncbi.nlm.nih.gov/projects/SNP/). Human Genome Build 36.3 (hg18) was used. The cDNA library was generated from total RNA isolated from transformed lymphoblastoid cells (RNeasy, Qiagen, Valencia, CA, USA; and Superscript III First-Strand Synthesis System, Invitrogen, Carlsbad, CA, USA). Amplification and sequencing of the DKC1 mRNA was performed as described.4
Linkage analysis
Genomic DNA was genotyped using the Infinium II Human linkage 12 panel (Illumina, San Diego, CA, USA). Single nucleotide polymorphisms (SNPs) used in the analysis had >99% call rates. The DeCode genetic map was used to describe SNP position. No SNPs were found to be out of Hardy–Weinberg equilibrium, and we used Pedcheck to confirm pedigree relationships and check for Mendelian inconsistencies.22 Linkage disequilibrium (LD) blocks (D'=0.7) were identified using Haploview,23 and tagging SNPs from each haplotype block were selected for subsequent linkage analysis to ensure the SNPs were not correlated. Linkage analysis was performed using MINX software using a parametric X-linked recessive model (prevalence of 0.0001 and penetrance of 0.0001, 0.05, 0.99 for females and penetrance of 0.0001, 0.99 for males).24
Western blot
Total protein was isolated from early passage lymphoblastoid cell pellets (passage 1–5) which were lysed in radioimmunoprecipitation assay (RIPA) buffer for 15 min on ice in the presence of protease inhibitors (Mini Complete, Roche, Indianapolis, IN, USA). Samples were centrifuged at maximum speed for 10 min, and total cellular protein quantitated using Bicinchoninic Acid assay (Pierce, Rockford, IL, USA). Proteins were separated using SDS-gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membrane under reducing conditions (Invitrogen). Primary antibodies for dyskerin and actin were obtained from Santa Cruz (sc-48794, lot D0809, Santa Cruz, CA, USA) and AbCam (ab8226, Cambridge, MA, USA), respectively. Detection was performed using near-infrared fluorescent conjugated secondary antibodies (LI-COR, Lincoln, NE, USA). We visualised the two proteins simultaneously on a single membrane without stripping using the two channel near infrared Odyssey scanner (LI-COR). We measured dyskerin protein levels by normalising dyskerin intensity to the intensity of the β-actin loading control using Odyssey software.
Quantitative reverse transcription PCR
Total RNA was isolated from early passage lymphoblastoid cells and reverse transcribed using Superscript III and random hexamers according to the manufacturer's protocol (Invitrogen). Quantitative reverse transcription PCR (qRT-PCR) was performed using Syber-Green Supermix with 200 nM final primer concentrations and 5–10 ng of cDNA. All products were cloned (TOPO TA cloning kit, Invitrogen) and sequence verified. Studies were performed in triplicate, and each experiment was performed at least twice. The expression in each sample was normalised to ARF3 levels.25 hTR was quantified by qRT-PCR.25 We quantified U23 and U3 levels as described.26 U64 forward and reverse primers were as follows: 5′-CTCGGCTCTGCATAGTTGC-3′ and 5′-CAAGGAAAGAGAGGCCACAG-3′. DKC1 expression was quantified using two primer sets. Primer set 1 spanned exons 11 and 12: 5′-CCCCGCTCTCCAGACCAGCT-3′ and 5′-AAGCCCCGCAGGTAGTTGCC-3′. Primer set 2 spanned exons 13 and 14: 5′-CTGCTATGGGGCCAAGATTA-3′ and 5′-CCATGGTCGCAGGTAGAGAT-3′.
Results
DC presents as familial pulmonary fibrosis
In patients with classic DC, 20% of patients have a diagnosis of pulmonary disease.1 To determine whether DC cases can be occult in a familial pulmonary fibrosis registry, we queried 75 consecutive probands for a history of oral leukoplakia, nail dystrophy or skin hyperpigmentation. We identified one proband who had classic DC features. The proband presented with shortness of breath at the age of 49 years and, at the time, had mild nail dystrophy and skin hyperpigmentation. Family history revealed a cousin who died from pulmonary fibrosis at the age of 39. This relative also had nail dystrophy, skin hyperpigmentation, and liver cirrhosis. These observations indicated that DC can manifest as familial pulmonary fibrosis, albeit rarely.
To determine the genetic basis of the DC phenotype in this family (designated #58), we screened genomic DNA from the proband for mutations in hTR and hTERT, the known genes that cause both familial pulmonary fibrosis and DC,19 27 but did not identify a mutation. The sequence for TINF2 was also intact. To probe the genetic basis further, we obtained a comprehensive five generation history. The family reported Irish ancestry, and the pedigree revealed premature mortality in males and three generations of individuals who had the typical mucocutaneous features of DC (figure 1, supplementary table 3). In total, the family history revealed eight males who died prematurely at a mean age of 37 years (range 17–51 years). Three died from complications of pulmonary disease alone, and three had both lung disease and liver cirrhosis. Another male died from metastatic squamous cell carcinoma at the age of 34 years (supplementary table 3). The family history did not reveal any cases of aplastic anaemia, myelodysplastic syndrome, or acute myeloid leukaemia. The pedigree suggested that this kindred may display X-linked recessive inheritance.

Pulmonary fibrosis pedigree with dyskeratosis congenita (DC) features. The proband is indicated by an arrow, and * indicates subjects from whom DNA was available. Individuals were assigned affected status if they had classic features of DC or very short telomeres (<1st centile). Females were assigned carrier status if they had affected male offspring or had skewed inactivation of the X-chromosome.
DC predominately affects males even in autosomal dominant families.28 To confirm the pattern of inheritance, we examined X-inactivation patterns using the HUMARA PCR based assay.21 We found skewed X-inactivation in genomic DNA from peripheral blood in two females (90% and 100% respectively, figure 2A). The skewing was also present in DNA extracted from saliva, consistent with a germline defect on the X-chromosome (figure 2B). Since females in X-linked DC have been previously described to have skewed X-inactivation,29 30 these data confirmed that the mutant gene in this family was on the X-chromosome.

Skewed X-inactivation in family 58 females and very short telomeres in affected individuals. (A) X-inactivation analysis of the androgen receptor locus in blood genomic DNA shows skewing in two females (IV.8 and IV.10, figure 1) compared to unrelated female controls. As expected, only a single allele is detected in genomic DNA from a male. Allele frequencies are compared with and without digestion with methylation specific restriction enzyme (HpaII) before PCR amplification. (B) X-inactivation analysis of genomic DNA from saliva from the same females also shows skewed ratios. (C) Lymphocyte telomere length of affected individuals are plotted by age relative to per cent normogram of healthy controls (n=400). Affected males have very short telomeres, at or less than the 1st centile, compared with age matched individuals. Carrier females also have telomere length near the 10th centile. Identifiers refer to pedigree position in figure 1. Males with known missense mutation Q31E also have very short telomeres by flow fluorescence in situ hybridisation (FISH).
Dyskerin sequence is unaltered in family 58
The DKC1 gene is mutated in X-linked DC3; we therefore sequenced its 15 coding exons in the proband but did not identify variants. Since the disease gene was presumed haploid on the X-chromosome, this analysis also indicated that the proband did not carry a deletion in DKC1. To exclude a mutation that altered splicing, we generated lymphoblastoid lines from an affected male and amplified the DKC1 cDNA. We identified no evidence of aberrant splicing or exon duplication. Therefore, the mutation that underlies inheritance in this X-linked DC family was not in the coding sequence and did not affect DKC1 splicing.
Affected individuals have short telomeres
To confirm whether the clinical syndrome in this X-linked DC family is telomere mediated, we measured the telomere length. Lymphocyte telomere length identifies patients with germline defects due to telomerase dysfunction.19 31 32 In four affected males, the telomere length was at or below the 1st centile compared to healthy age matched controls (p=0.009, paired t test, figure 2C). This range, which is three standard deviations below the mean, is diagnostic of an inherited mutation that affects telomerase or telomere maintenance. In a healthy male sibling, the telomere length was greater than the 50th centile (figure 2C). These data suggested that the inherited defect in this DC family affects telomere maintenance.
Genome wide linkage identifies the DKC1 locus
The absence of a readily identifiable mutation in the DKC1 gene and the presence of short telomeres suggested two main possibilities: either another gene on the X-chromosome which affects telomere maintenance underlies the inheritance in this family, or the DC features in this family can be attributed to a novel mechanism which affects dyskerin function. To begin to distinguish these, we performed linkage analysis on the X chromosome using SNP data from 16 individuals. Using an X-linked recessive model, we identified a single 47 cM peak on Xq28 with a maximum log of the odds ratio (LOD) score of 3.25 (figure 3, supplementary table 4). The remainder of the X chromosome was not involved as the LOD score was less than −2. This linkage peak flanked and contained the genomic locus for dyskerin (figure 3). We examined bioinformatically the 134 protein coding genes that fell within the linkage peak and found that only DKC1 had been previously implicated in telomere maintenance. However, several genes of unknown function were also present under the linkage peak. A list of the protein coding genes is listed in supplementary table 5. Therefore, although the DKC1 sequence was intact, an inherited mutation affecting the DKC1 locus was responsible for the inheritance of the disease phenotype.
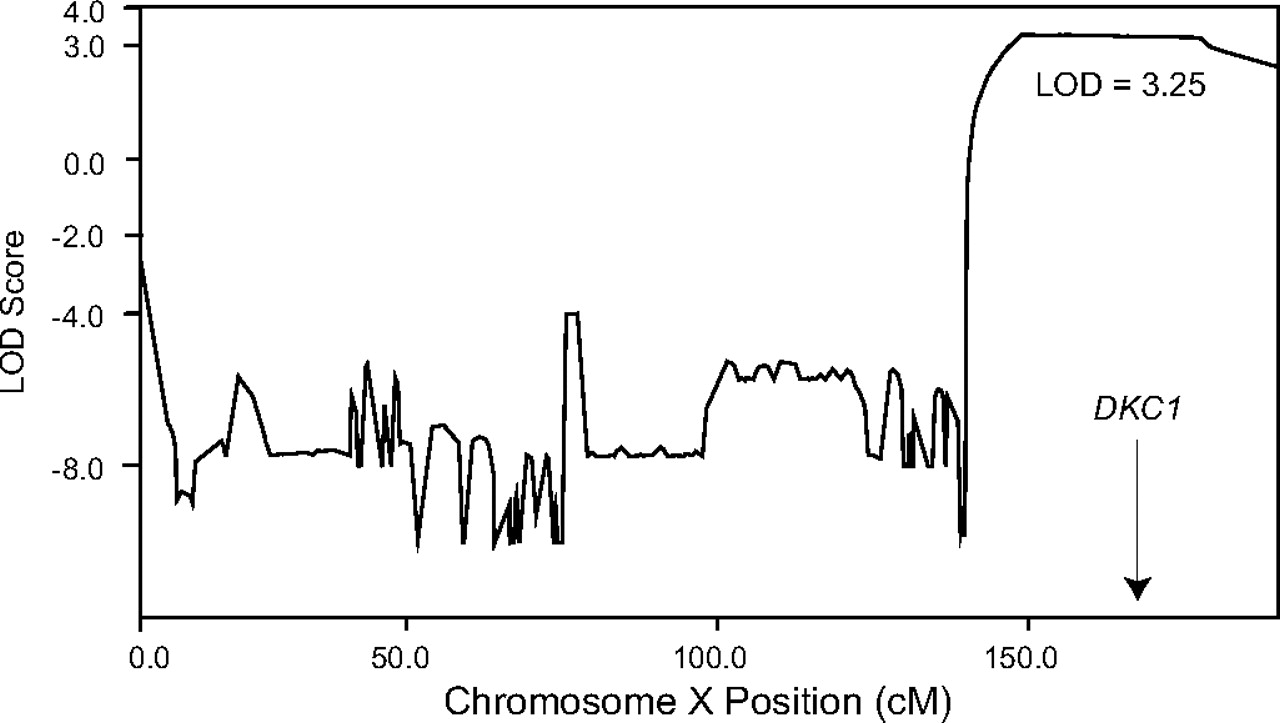
X-chromosome linkage analysis points to the DKC1 locus. Log of the odds ratio (LOD) score plot across the X-chromosome demonstrates linkage to Xq with a maximum LOD score of 3.25. The linkage peak includes the gene for dyskerin, DKC1, at Xq28.
Affected individuals have reduced telomerase RNA levels
Dyskerin is a box H/ACA protein and is essential for hTR stability and biogenesis.5 7 In patients with point mutations in DKC1, low hTR levels cause telomere shortening and telomere mediated disease.7 Since our linkage study suggested that a mutation that affects DKC1 may underlie the inheritance in this DC family, we tested whether affected individuals have low hTR levels. We quantified hTR in cells from affected males and compared them to healthy controls using quantitative real time PCR. We found hTR levels were 25% of control levels (p=0.043, Student t test, figure 4A). hTR levels were also decreased in cells from DC patients who carried a known missense dyskerin mutation (Q31E), as previously shown.8 We examined but did not detect a difference in U23 and U64, two H/ACA snoRNAs. There was also no significant difference in the levels of U3, a member of the box C/D snoRNA family (figure 4B). These data suggested that the primary defect in this family causes telomere shortening, by decreasing hTR stability.

Telomerase RNA and dyskerin protein levels are compromised in affected males. (A) Quantitative reverse transcription PCR (qRT-PCR) of hTR shows decreased levels in affected males compared with controls. hTR levels are also decreased in cells with a dyskerin Q31E missense mutation, as was previously shown. (B) Quantitation of snoRNAs (two box h/ACA and one box C/D) shows no change between affected individuals and controls. qRT-PCR data were normalised relative to ARF3 levels. (C) Representative grey scale Western blot image of one of the near-infrared colour fluorescent images used for quantitation. Molecular weight standard is shown on the left (kDa). Dyskerin appears at the expected size of 58 kDa in affected individuals designated with ‘A’ and controls designated ‘C,’ as well as Hela cells. Lane 10 contains protein extract from a healthy male from family 58 who has long telomeres (pedigree ID V.3, figure 1). A total of 10 μg of protein was loaded in each lane. (D) Quantitation of near-infrared fluorescence intensity from five independent experiments including the gel shown in (C) shows decreased dyskerin levels in affected males compared with controls. Each experiment contained control cells (n=5–7), affected individuals (n=3–4), and Q31E mutation carriers (n=2). Studies shown here used RNA and protein extracted from early passage lymphoblastoid lines (passage 1–5). Means were compared using the Student t test. Error bars represent SE of the mean, and p values are two sided.
Dyskerin protein levels are decreased
Since the linkage analysis along with decreased hTR levels suggested that the DKC1 gene locus contained the culprit defect, we tested whether a decrease in dyskerin protein levels may underlie the telomere shortening in this pedigree. We measured dyskerin levels by Western blot using a sensitive near-infrared fluorescence detection method.33 Total protein extracted from lymphoblastoid cells from clinically affected members contained the expected 58 kDa product identical to Hela cells and controls (representative image, figure 4C). However, in males who had short telomeres, dyskerin protein levels were significantly decreased relative to actin. In five independent experiments, we detected a decrease in dyskerin levels with a mean 67% of controls (range 52–72%, p<0.0001, Student t test, figure 4D). The dyskerin protein levels in males were also lower compared to a healthy related male with long telomeres (figure 4C). Our data indicated that decreased dyskerin protein levels contributed to the telomere shortening in this family.
Examining DKC1 transcriptional regulatory elements
The decrease in dyskerin protein levels in the presence of an intact coding and cDNA sequence raised the possibility that a mutation that might affect proximal DKC1 regulatory elements underlies the genetic defect. However, quantitation of DKC1 expression using two independent primer sets was intact (supplementary figure 1). Nonetheless, to test this possibility, we amplified the 5′ upstream sequences which include the promoter (0.8 kb), as well as the 3′ UTR (0.8 kb), but found no variants. To determine whether regulatory elements may be located near dyskerin, we aligned mammalian DKC1 sequences. Based on conservation algorithms available (phastCons, http://genome.ucsc.edu), we found no conserved elements in non-coding regions, and prediction programs did not identify putative enhancers or transcription factor binding sites (Vista Enhancers and Yale TFBS, http://genome.ucsc.edu, supplementary figure 2). Insulator prediction programs predicted an insulator element within intron 1 (http://insulatordb.uthsc.edu/), but sequencing of both introns 1 and 2 did not reveal variants from reference. The sequences of snoRNAs (SNORA36a and SNORA56) within introns 8 and 12 were also intact. To determine if more remote elements were involved, we amplified and sequenced the 11.6 kb upstream and 1 kb downstream intergenic sequences (supplementary figure 2), but found no deviations from reference sequence with the exception of a G to A transition, 7.8 kb upstream of dyskerin, within a long interspersed nuclear element (LINE). This is the only sequence variant we identified in the proband near the DKC1 gene. We also considered the possibility that a balanced translocation that disrupted the regulatory element may underlie the phenotype in this family, but metaphase FISH with X-chromosome specific probes did not reveal a micro-translocation. These data suggested that a micro-translocation larger than the resolution of metaphase FISH probes (1–3 Mb) is not likely.34 Altogether, the intact expression of DKC1, along with the absence of readily identifiable variants in known regulatory elements, suggested that a potentially novel mechanism that controls dyskerin protein levels is defective in this family.
Carrier females have short telomeres and mild DC features
We next examined the clinical phenotypes in carrier females where attenuated disease features are known to occur in X-linked disorders.35 We identified nail ridging characteristic of DC in one of four female carriers examined (figure 5A). Female carriers also had telomere lengths that were shorter than the 50th centile of controls (n=4, p=0.022, paired t test, figure 2C). Since telomere length is inherited,36 these data suggested that telomere shortening may contribute to the low penetrance dyskeratosis phenotype in some carrier females.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
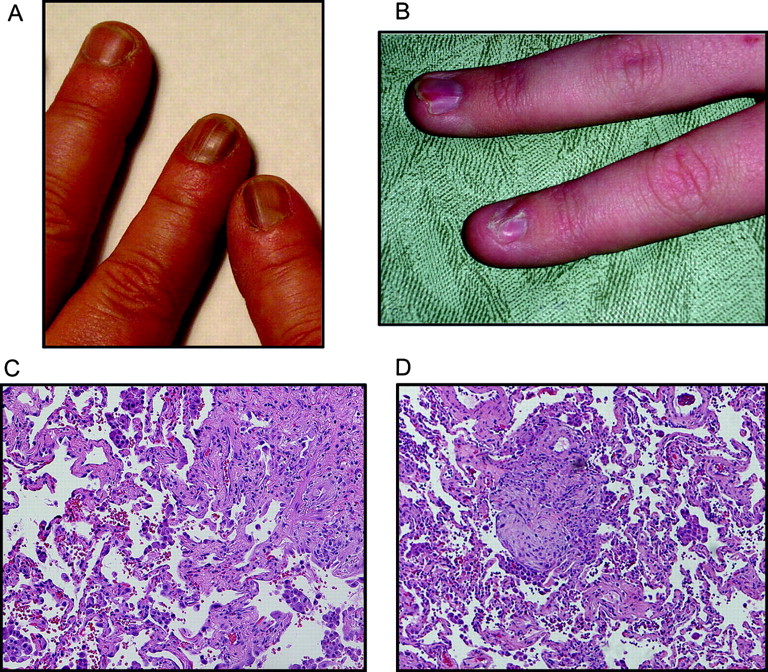
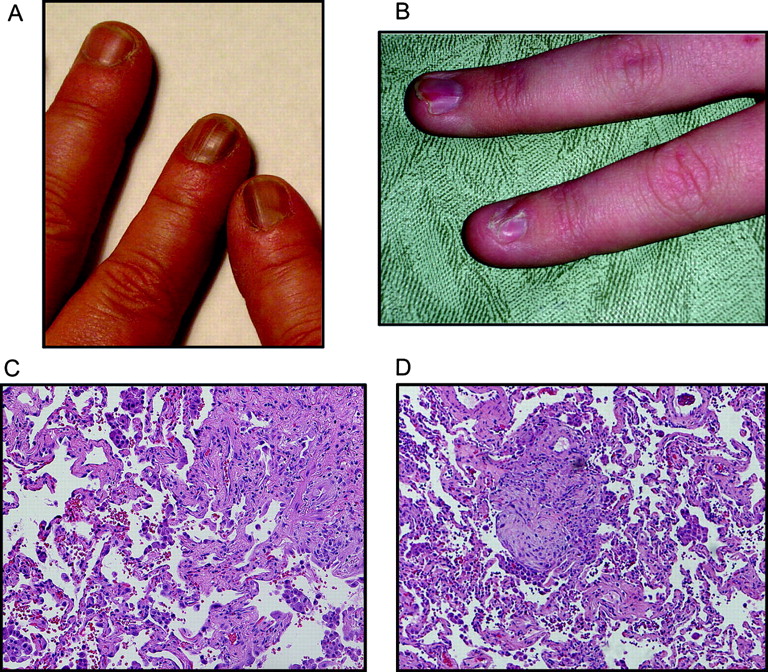
Clinical features of affected members. (A) Dystrophic nail changes with typical ridging of dyskeratosis congenita (DC) are shown in this photomicrograph of a female carrier. The telomere length for this individual is shown in figure 2C. (B) Nail dystrophy in an affected male child with very short telomeres in figure 2C. (C) Lung biopsy from the proband shows changes of bronchiolitis obliterans organising pneumonia and usual interstitial pneumonia patterns typical of idiopathic pulmonary fibrosis. (D) Biopsy from another affected individual (III.10) shows late stage idiopathic pulmonary fibrosis changes of bronchiolitis obliterans organising pneumonia and usual interstitial pneumonia. Subject gave informed consent for this study which included permission to publish clinical images.
Prominent pulmonary and liver phenotypes in DC kindred
To determine the spectrum of telomere mediated disease associated with decreased dyskerin levels, we carefully examined the clinical phenotype. In several young males, nail dystrophy was the first manifestation and was evident by the age of 15 (figure 5B and supplementary table 3). In the two cases where pulmonary fibrosis was biopsy proven, there was evidence of usual interstitial pneumonia as well as bronchiolitis obliterans organising pneumonia, a histologic manifestation which sometimes accompanies idiopathic pulmonary fibrosis (figure 5C,D). Both patients had honeycomb changes radiographically, did not respond to steroids, and died from their lung disease. These cases confirm prior observations suggesting that other interstitial lung disease histopathologies may co-exist in the same individual or within the same family along with usual interstitial pneumonia, and are manifestations of the same underlying genetic process.19 37 Although aplastic anaemia and cytopenias occur in >85% of patients with classic features of DC,1 38 a detailed medical records review from 12 affected individuals did not reveal evidence of bone marrow failure. The data suggested that patients with classic DC can have their most prominent complications in the lung, and that these manifestations are sufficient to cause life threatening disease in the absence of bone marrow failure.
Discussion
In this study, we identify decreased dyskerin protein levels as a novel mechanism of disease in X-linked DC. In affected individuals, despite the presence of an intact coding sequence, decreased dyskerin levels were associated with reduced hTR stability and telomere shortening. Knockdown of dyskerin levels has been previously shown to decrease hTR levels in cancer cell lines,39 40 although its consequences on telomere length have not been examined. Our data indicate that intact dyskerin levels, and not only protein sequence, are essential for in vivo telomere maintenance, and this defect is sufficient to cause premature mortality due to telomere mediated disease. In half of X-linked DC families, the mechanism of disease is not known. Our data, if replicated in larger cohorts, would suggest that quantitation of dyskerin protein levels can complement sequencing of the DKC1 gene, and decreased levels of dyskerin, measured by sensitive methods, can identify male carrier status in a subset of genetically uncharacterised families.
Our family study highlights the high penetrance of pulmonary disease in patients with classic features of DC. Traditionally, DC has been associated with premature mortality from bone marrow failure.41 42 Recent studies showing that mutations in hTERT and hTR have their most common manifestations in the lung (reviewed by Armanios13), along with the findings in this study, suggest that the penetrance of pulmonary phenotypes in families with DC may have been underestimated. In a DC registry where probands were recruited based on the classic mucocutaneous features, 20% of patients had pulmonary disease.1 Our study indicates that, in the absence bone marrow failure, pulmonary and liver disease can be primary causes of mortality in some DC families. Similar to autosomal dominant families with mutations in hTERT or hTR, the median age of death due to pulmonary disease was older (39 years), nearly two decades later than bone marrow failure onset in DC registries.19 41 These observations underscore the heterogeneity of DC phenotypes, and the fact that pulmonary disease may represent an attenuated, adult onset telomere phenotype which manifests rarely in individuals younger than the age of 20 years. These patterns have important clinical implications as DC patients should be rigorously counselled against tobacco exposure, and iatrogenic pulmonary toxins should be avoided because of the known high rate of complications.43 Although in our study female carriers were clinically unaffected except for the presence of nail dystrophy, it is possible the short telomeres they carry may be associated with an increased risk of developing telomere mediated disease, especially later in life. Future studies are needed in this area.
How do we explain the observation that, in this family, affected males had decreased dyskerin protein and hTR levels, yet no change in DKC1 mRNA levels? Several possibilities may account for this. MicroRNAs (miRNAs) are post-transcriptional regulators of protein levels.44 It is theoretically possible that this family harbours a gain-of-function miRNA mutation that accounts for this observation. Disease causing germline mutations in miRNAs have been hypothesised, but to our knowledge, have not yet been shown to cause Mendelian disease.45 In the linkage peak, there are 32 predicted miRNAs genes (listed in supplementary table 5) which are potential candidates. Regulatory RNAs known as large intergenic non-coding RNAs (lincRNAs) have also been recently shown to regulate protein levels directly, and it is possible that a mutation that affects these genes would alter dyskerin levels.46 No lincRNAs have been heretofore identified within our linkage peak. Another alternative explanation is that a loss-of-function mutation in a protein coding gene that has a yet-to-be identified function in telomere biology underlies the decreased dyskerin levels. Several biochemical studies have examined the dyskerin interacting proteins,47–49 but none of them are identifiable as one of the protein genes that fall under the linkage peak in this family. Examining these possibilities in future studies could lead to important insights into the relevant genetic determinants of telomere length and age related disease in humans.
Although we identified significant decreases in hTR associated with lower dyskerin levels, we did not identify a compromise in the H/ACA snoRNA levels we examined. Our data are similar to prior studies which did not identify decreases in snoRNA levels in cells from patients with DKC1 missense mutations.7 8 These data differ from those in mouse cells where DKC1 mutations were associated with a decrease in snoRNA levels.9–11 The contrast between human and mouse studies suggests that in human cells, hTR levels may be more sensitive to dyskerin dose fluctuation than snoRNAs.
In summary, intact dyskerin levels are critical for hTR stability and in vivo telomere maintenance. A decrease in dyskerin protein, in the absence of coding mutations, is associated with a spectrum of telomere mediated disease which clinically manifests as familial pulmonary fibrosis and DC. If replicated in other families, careful quantitation of dyskerin protein levels may be useful for identifying male carrier status in uncharacterised X-linked DC families.
Acknowledgments
We are grateful to all the family members who participated in this study. This work was supported by funding from the National Institutes of Health (K08CA118416) and the Doris Duke Charitable Foundation (to MA). EMP received support from an NHLBI supplement to Medical Scientist Training Program Award T32GM007309.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Funding Other Funders: NIH; Doris Duke Foundation.
Patient consent Obtained.
Ethics approval Approval was provided from both the Johns Hopkins and Vanderbilt University School of Medicine's Institutional Review Boards.
Provenance and peer review Not commissioned; externally peer reviewed.