Article Text

Abstract
Background Congenital diaphragmatic hernia (CDH) is a life threatening birth defect. Most of the genetic factors that contribute to the development of CDH remain unidentified.
Objective To identify genomic alterations that contribute to the development of diaphragmatic defects.
Methods A cohort of 45 unrelated patients with CDH or diaphragmatic eventrations was screened for genomic alterations by array comparative genomic hybridisation or single nucleotide polymorphism based copy number analysis.
Results Genomic alterations that were likely to have contributed to the development of CDH were identified in 8 patients. Inherited deletions of ZFPM2 were identified in 2 patients with isolated diaphragmatic defects and a large de novo 8q deletion overlapping the same gene was found in a patient with non-isolated CDH. A de novo microdeletion of chromosome 1q41q42 and two de novo microdeletions on chromosome 16p11.2 were identified in patients with non-isolated CDH. Duplications of distal 11q and proximal 13q were found in a patient with non-isolated CDH and a de novo single gene deletion of FZD2 was identified in a patient with a partial pentalogy of Cantrell phenotype.
Conclusions Haploinsufficiency of ZFPM2 can cause dominantly inherited isolated diaphragmatic defects with incomplete penetrance. These data define a new minimal deleted region for CDH on 1q41q42, provide evidence for the existence of CDH related genes on chromosomes 16p11.2, 11q23-24 and 13q12, and suggest a possible role for FZD2 and Wnt signalling in pentalogy of Cantrell phenotypes. These results demonstrate the clinical utility of screening for genomic alterations in individuals with both isolated and non-isolated diaphragmatic defects.
- Diaphragmatic hernia
- ZFPM2
- microdeletion 1q41q42
- microdeletion 16p11.2
- FZD2
- clinical genetics
- cytogenetics
- molecular genetics
Statistics from Altmetric.com
- Diaphragmatic hernia
- ZFPM2
- microdeletion 1q41q42
- microdeletion 16p11.2
- FZD2
- clinical genetics
- cytogenetics
- molecular genetics
Introduction
Congenital diaphragmatic hernia (CDH) is a life threatening birth defect that occurs in approximately 1/4000 live births; it is defined as a protrusion of abdominal viscera into the thorax through an abnormal opening or defect that is present at birth.1 2 In some cases the herniated viscera are covered by a membranous sac. This type of CDH can be difficult to distinguish from a diaphragmatic eventration in which there is extreme elevation of a part of the diaphragm that is often atrophic and abnormally thin. The majority of CDH cases are sporadic, with the recurrence risk for isolated CDH typically quoted as <2% based on a model of multifactorial inheritance.3 However, there is a growing body of evidence that small chromosomal anomalies can predispose to the development of CDH.4–10
The identification of a predisposing genomic change greatly enhances the ability of physicians to provide individualised genetic counselling and to formulate optimal treatment and surveillance plans. This would suggest that screening for genomic changes by array comparative genomic hybridisation (aCGH) or single nucleotide polymorphism (SNP) based copy number analysis should be performed on all patients with diaphragmatic defects.10 11 However, this practice has not been universally applied.
Efforts to map the locations of all reported chromosomal anomalies associated with CDH have revealed many regions of the genome that are recurrently deleted, duplicated, or translocated in individuals with CDH.12 13 Each of these genomic regions is likely to harbour one or more CDH related genes. For some of theses regions, a specific gene has been implicated in the development of diaphragmatic defects. Chromosome 8q22q23, for example, is recurrently deleted and translocated in individuals with CDH. This region harbours the zinc finger protein, multitype 2 (ZFPM2) gene. ZFPM2 has been implicated in the development of diaphragmatic defects, based the identification of a de novo truncating mutation in a child with a severe diaphragmatic eventration and the development of similar diaphragmatic defects in mice that are homozygous for a hypomorphic allele of Zfpm2.14 In contrast, chromosome 1q41q42—which is also recurrently deleted in individuals with CDH—harbours a number of genes, none of which have been clearly shown to cause diaphragmatic defects in humans or animal models.15
In this report we screened a cohort of 45 individuals with diaphragmatic defects for genomic alterations by high resolution aCGH or SNP based copy number analysis. Chromosomal anomalies that are likely to have caused or contributed to the development of diaphragmatic defects were identified in eight patients. These findings allow us to define the inheritance pattern of diaphragmatic defects associated with haploinsufficiency of ZFPM2 and delineate a new minimal deleted region for CDH on 1q41q42. They also provide evidence for the existence of CDH related genes on chromosomes 16p11.2, 11q23-24 and 13q12 and suggest a possible role for FZD2 and Wnt signalling in the development of various phenotypes seen in pentalogy of Cantrell.16
Methodology
Patient accrual
For array studies, informed consent was obtained from a convenience sample of 45 patients with CDH or diaphragmatic eventrations and, when possible, their parents in accordance with institutional review board approved protocols. Clinical and molecular data from these patients have not been published with the exception of patients 5 and 7 whose clinical findings were summarised by Shinawi et al and Fruhman et al, respectively.17 18
For sequence analysis of ZFPM2, a cohort of 52 patients with CDH and 10 patients with diaphragmatic eventrations were screened for deleterious changes. This cohort consisted of a subset of patients from the array cohort and similarly consented individuals. ZFPM2 sequence analyses for these individuals have not been previously published.
aCGH and SNP based copy number analyses
In most cases, chromosomal deletions and duplications were identified or confirmed by aCGH on a research basis using Human Genome CGH 244K or SurePrint G3 Human CGH 1M Oligo Microarray Kits (G4411B, G4447; Agilent Technologies, Santa Clara, California, USA), prepared according to the manufacturer's protocols and analysed as previously described using individual sex matched controls with no personal or family history of CDH.7 Putative copy number changes were defined by two or more adjacent probes at 244 K resolution or three or more adjacent probes at 1 M resolution with log2 ratios suggestive of a deletion or duplication when compared to those of adjacent probes.
In two cases—patients 2 and 6—causative chromosomal deletions were identified before accrual and further aCGH testing was unwarranted based on the molecular data already available. The deletion in patient 2 was identified using an Illumina CytoSNP bead version 12.2 (Illumina, Inc, San Diego, California, USA) and the deletion in patient 6 was identified and defined on a clinical basis using a 105 K Combimatrix Molecular Diagnostics array (Combimatrix Molecular Diagnostics, Irvine, California, USA) hybridised, extracted, and evaluated according to manufacturer's instructions.
Identification of previously reported copy number variants
To determine if putative changes identified by aCGH or SNP based copy number analysis had been described previously in normal controls, we searched for similar deletions or duplications in the Database of Genomic Variants (http://projects.tcag.ca/variation/).
Confirmation of genomic changes
Changes that were not identified in the Database of Genomic Variants were confirmed by real-time quantitative PCR, with the exception of causative changes identified in patients 3 and 7 which were confirmed by chromosome analysis, and patients 4 and 6 which were confirmed by fluorescence in situ hybridisation (FISH) analysis.
Quantitative real-time PCR
Quantitative real-time PCR (qPCR) analysis experiments were designed and carried out as previously described.7 For qPCR analysis within the ZFPM2 gene, experiments were designed in a manner similar to the standard curve method described by Boehm et al with a region of the c14orf145 gene serving as a control locus.19
Chromosome analyses and FISH studies
Chromosome analyses were performed for patients 3 and 7 on a clinical basis by the Molecular Genetics Laboratory at Baylor College of Medicine. FISH analyses were performed for patient 4 by the Cytogenetic Laboratory at Texas Tech University Health Sciences Center School of Medicine and for patient 6 by Combimatrix Molecular Diagnostics.
Long range PCR amplification and sequencing
Long range PCR amplification was carried out using the TaKaRa long range PCR system (TaKaRa Bio, Otsu, Shiga, Japan) according to manufacturer's instructions. PCR products were gel purified, sequenced, and analysed using Sequencher 4.7 software (Gene Codes Corporation, Ann Arbor, Michigan, USA).
Identification of ZFPM2 sequence changes
Primers were designed to amplify the coding sequence and the intron/exon boundaries of ZFPM2 and used to amplify patient DNA. Sequence changes in PCR amplified products were identified by comparison with control DNA sequences using Sequencher 4.7 software (Gene Codes Corporation, Ann Arbor, Michigan, USA).
Hispanic control samples were obtained from the Baylor Polymorphism Resource, a collection of approximately 600 anonymous control samples from major ethnic and racial backgrounds.
Results
Identification of genomic changes in patients with CDH or diaphragmatic eventrations
Forty-five subjects with congenital diaphragmatic hernias or diaphragmatic eventrations of varying severity were screened for chromosomal anomalies by aCGH or SNP based copy number analysis. Eight subjects carried genomic changes that likely contributed to the development of their diaphragmatic defects. Clinical and molecular data from these subjects are summarised in table 1.
Summary of data from subjects with chromosomal changes likely to have contributed to diaphragm defects
Patient 1 is a male of mixed ancestry who was diagnosed at 9 days of age with intestinal malrotation and a large, left-sided diaphragmatic eventration (figure 1). Later in life, he was diagnosed with left-sided radioulnar synostosis. Despite normal motor development, at 24 months of age he spoke only two words. However, he made rapid progress with speech therapy and by 31 months of age he was speaking in four-word sentences.

(A) Array comparative genomic hybridisation (aCGH) data from patient 1 showing an 8q22.3q23.1 single gene deletion of ZFPM2. (B) The relative location of the ZFPM2 gene in relation to aCGH data from the deletion region in patient 1 is represented by a grey bar. (C) Chest radiograph of patient 1 demonstrating a severe diaphragmatic eventration containing loops of bowel. (D) The same radiograph shown in panel C with the limits of the diaphragmatic eventration outlined in yellow. (E) Quantitative PCR analysis demonstrates a normal copy number for ZFPM2 exon 6 but a reduced copy number for exons 7 and 8 in DNA from patient 2 (Pt2) and his mother (M). Normal copy number values are seen in DNA from patient 2′s father (F) and DNA from two unrelated controls (C1, C2).
aCGH analysis revealed an ∼1 Mb deletion that included only the ZFPM2 gene on chromosome 8q22.3-23.1 (figure 1). Quantitative and long range PCR analysis revealed that the patient's father carried the same deletion (data not shown). A review of his father's medical records did not reveal evidence of a diaphragmatic anomaly despite an extensive evaluation after a severe automobile accident which included an abdominal CT scan. We screened the coding sequence and associated intron/exon boundaries of patient 1's remaining ZFPM2 allele looking for changes that might account for the difference in phenotype between him and his father, but no sequence changes were identified.
Patient 2 is a Caucasian male that was diagnosed prenatally with a left-sided CDH. His brother was similarly affected with CDH and died of complications related to his hernia. Except for a single umbilical artery, no additional anomalies were identified after birth. He died at 2 days of age from respiratory insufficiency and pulmonary hypertension. SNP based copy number analysis revealed a maternally inherited deletion on chromosome 8q23.1 that included a portion of the 3′ coding region of ZFPM2 and the first coding exon of oxidation resistance gene 1 (OXR1) based on mRNA transcript variant 1. Quantitative PCR analysis revealed that exons 7 and 8 of ZFPM2 were included in the deletion (figure 1). No sequence changes were identified in the coding sequence and intron/exon boundaries of the remaining allele.
Patient 3 had a de novo ∼32 Mb deletion from 8q22.3 to 8q24.23 that overlapped the ZFPM2 deletions seen in patients 1 and 2 and also included the EXT1 and TRPS1 genes that are associated with Langer–Giedion syndrome (OMIM #150230). This Hispanic female was diagnosed prenatally with a large left-sided posterolateral CDH. Fetal MRI at 34 5/7 weeks also revealed an omphalocele and symmetrically short extremities. Due to severe pulmonary hypoplasia she was placed in neonatal hospice care immediately after birth and died shortly thereafter.
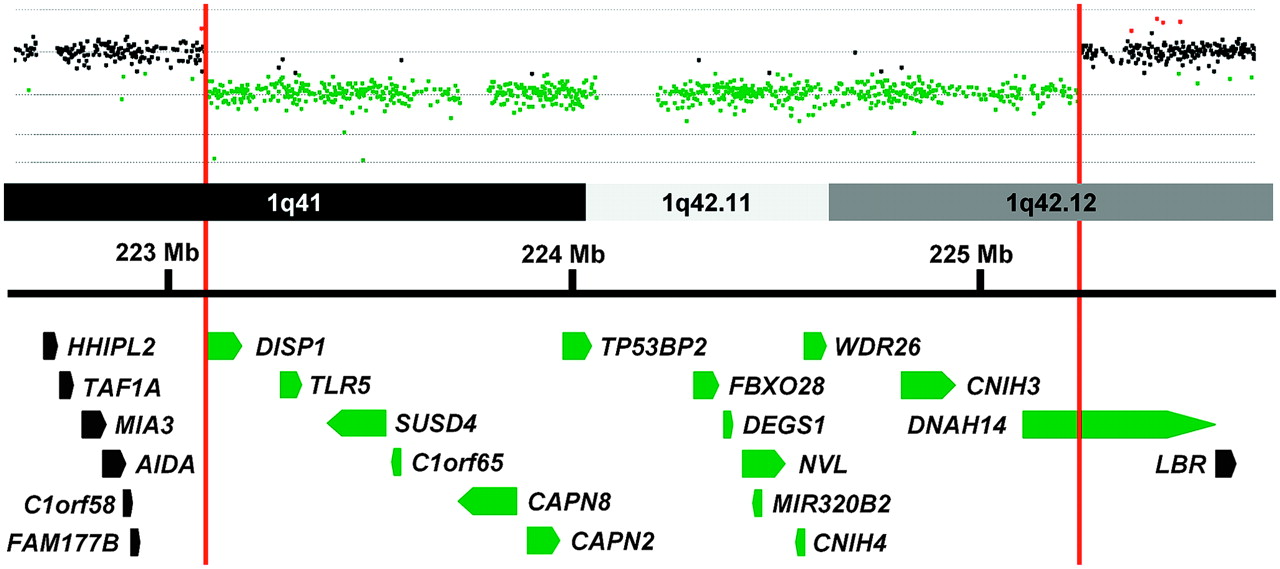
Patient 4 is a Caucasian male who had a normal prenatal course. He was born at 37 weeks gestation and his perinatal course was complicated by persistent hypoglycaemia. A diaphragmatic hernia was identified incidentally on a chest x-ray at 11 months of age during an investigation for failure to thrive. His motor and language development were delayed and he developed seizures at 20 months of age. Central nervous system (CNS) studies revealed right hemisphere cerebral volume loss with ex vacuo dilatation of the right lateral ventricle with diffuse thin cortical mantle, atrophy in the right hippocampus, and a thin corpus callosum. aCGH analysis revealed a de novo 2.2 Mb interstitial deletion of 1q41-1q42.12 (figure 2).

{kind=link}
{kind=link}
Array comparative genomic hybridisation data from patient 4 is shown above a schematic representation of a portion of the 1q41-1q42 region. Genes in this region are represented by block arrows. Red vertical bars mark the limits of the deletion in patient 4 and delineate the minimal deleted region for congenital diaphragmatic hernia (CDH) in this region of the genome. Genes with all or a portion of their coding sequence inside the minimal deleted region are shown in green.
Patients 5 and 6 were found to have de novo interstitial deletions of 16p11.2—a recurrent microdeletion region flanked by low copy repeats. Patient 5 is a 2-year-old male of mixed ancestry who had a severe right-sided posterolateral CDH, micrognathia, a U-shaped palatal cleft, paternally inherited autosomal dominant polydactyly, and dysmorphic features consistent with a 16p11.2 deletion which were previously described.17 He continues to need dietary supplementation via G-tube and supplemental oxygen when ill.
Patient 6 was a male infant conceived by intrauterine insemination with non-consanguineous Caucasian parents as donors. A left-sided diaphragmatic hernia was diagnosed prenatally. After birth, both of his thumbs were found to be proximally placed, hypoplastic and non-articulating, with the left thumb having a pedunculated appearance. Additional findings included an extra thoracic vertebra and 13 pairs of ribs. He required extracorporeal membrane oxygenation starting on the first day of life, developed severe diffuse oedema, and died on the 17th day of life as a result of his severe respiratory insufficiency and pulmonary hypertension.
Patient 7 was born at 37 6/7 weeks to non-consanguineous Hispanic parents. She had persistent respiratory distress at birth and intubation was complicated by micrognathia and a tongue anomaly. Further evaluation revealed a left-sided CDH, a ventricular septal defect (VSD) and atrial septal defect (ASD), partial cleft palate, small anteriorly placed anus, and dysmorphic features. A marker chromosome was identified by G-banded chromosome analysis and was shown be the result of an unbalanced maternal translocation between chromosome 13q and 11q, resulting in a 47,XX,+der(13)t(11;13)(q23;q12.3) chromosome complement.18 Her prognosis was deemed poor in light of her multiple anomalies, and she died shortly after support was withdrawn on day 8 of life. Additional findings at autopsy included abnormal lung fissures and coronary artery anomalies.
Patient 8 is a 2-year old Caucasian male who was diagnosed prenatally with a left-sided anterior CDH, and a large omphalocele. Postnatal cardiac evaluation revealed a large perimembranous VSD and a patent foramen ovale versus ASD. Additional anomalies included bilateral inguinal hernias, left-sided cryptorchidism, and premature fusion of the coronal sutures evident on head CT performed at 7 and 12 months of age. An MRI obtained at 11 months of age revealed diminished bifrontal and bitemporal parenchyma white matter associated with thinning of the corpus callosum, delayed myelination, and generous extra-axial cerebrospinal fluid (CSF) spaces.
aCGH analysis revealed a de novo 17.4 kb deletion that involved only the frizzled homologue 2 gene (FZD2) on chromosome 17q12.2. Amplification of the breakpoint by long range PCR followed by sequencing revealed that the deletion had occurred between two Alu repeats; an Alu-Sb proximally and an Alu-Sq distally. Matsuzaki et al has reported deletions of this gene in 3/90 normal Yoruban individuals from Ibadan, Nigeria.20 However, these changes were not identified by McCarroll et al who analysed the same cohort.21
To determine if the mutations in patient 8's remaining FZD2 allele may have contributed to his phenotype, we sequenced the coding region of his remaining FZD2 allele but did not identify any sequence changes.
Twenty changes not reported in the Database of Genomic Variants were identified in this cohort by aCGH and confirmed by either qPCR and/or FISH analysis (table 2). In two cases the extent of the copy number variation was larger than that seen in the parents. Patient TX-28 appeared to have four copies of a region on 11q13.5, while her parents each had three copies compared to control. Patient TX-50 had no copies of a region on 17q25.1, while his parents each had one copy compared to control. TX-50 also had two copies of a region on Xp27.1, similar to his mother, which could represent a de novo event.
Rare inherited changes identified in patients with congenital diaphragmatic hernia
Identifying ZFPM2 sequence changes in patients with CDH and diaphragmatic eventrations
To determine if detrimental sequence changes in ZFPM2 were a common cause of CDH or diaphragmatic eventrations, we screened the coding region and intron/exon boundaries of this gene in 52 patients with CDH (table 3) and 10 patients with diaphragmatic eventrations (table 4). Three non-synonymous changes were identified which had not been previously described in the 1000 Genomes project or dbSNP. A D98N change was identified in a male with an isolated posterior left-sided hernia with a large sac. His mother did not carry a D98N allele, but the father was unavailable for analysis. This change was predicted to be possibly damaging by PolyPhen (http://genetics.bwh.harvard.edu/pph/) but non-deleterious by SNPs3D (http://www.snps3d.org/).22 23 The same change was reported by Bleyl et al in an individual with bilateral CDH.24 However, they concluded that the change was likely to be a benign SNP since it was also identified in control samples.
ZFPM2 sequence changes in patients with congenital diaphragmatic hernia
ZFPM2 sequence changes in patients with diaphragmatic eventrations
A Q889E change was identified in a Hispanic female with a right-sided CDH, ambiguous genitalia, and a double vagina. Her mother did not carry the change, but the father was unavailable for analysis. This change was predicted to be benign by PolyPhen, non-deleterious by SNPs3D, and was subsequently identified in five of 152 (3.3%) Hispanic control chromosomes.
A paternally inherited S210T mutation was identified in an African American male with a left-sided CDH and dysmorphic features. This change was predicted to be benign by PolyPhen and non-deleterious by SNPs3D.
Discussion
aCGH and SNP based copy number analyses are commonly used as a primary screening tool to identify genomic aetiologies in patients with congenital anomalies. Identification of these changes can provide information that physicians can use to hone the prognostic and recurrence risk information given to families and to create individualised therapeutic and surveillance plans for patients.
Haploinsufficiency of ZFPM2 causes autosomal dominant isolated diaphragmatic defects with incomplete penetrance
ZFPM2 is a transcriptional co-factor that forms heterodimers with GATA transcription factors to regulate gene expression during development.25 The role of ZFPM2 in development of diaphragmatic defects was first demonstrated by Ackerman et al who showed that mice homozygous for a hypomorphic mutation in Zfpm2 develop diaphragmatic eventrations.14 In the same publication, they reported a de novo heterozygous R112X mutation in an infant with severe diaphragmatic eventrations and pulmonary hypoplasia who died shortly after birth.14
Patient 1 is the second child in which haploinsufficiency for ZFPM2 has been seen in association with a severe diaphragmatic eventration. Although patient 2′s deletion affects both the 3′ end of ZFPM2 and the first coding exon of OXR1, it is likely that disruption of ZFPM2 underlies the development of CDH in this patient since OXR1 appears to be involved in the prevention of oxidative damage—a process that has not been implicated in the development of diaphragmatic defects.26 Although DNA is not available on patient 2′s brother, it is likely that he inherited the same deletion from his mother which contributed to his CDH. Alterations in ZFPM2 activity may also be the major, if not the sole, genetic factor that contributed to the development of CDH in patient 3 and previously reported individuals with CDH with deletions and translocation involving 8q22q23 (supplemental table 2).13 Since both patients 1 and 2 inherited their deletions from unaffected parents we conclude that loss-of-function alleles of ZFPM2 are associated with autosomal dominant diaphragmatic defects—CDH or diaphragmatic eventrations—with incomplete penetrance.
The identification of the causative genomic changes in patients 1 and 2, both of whom had only minor anomalies—radioulnar synostosis diagnosed at 2 ½ years of age and a single umbilical artery, respectively—provide further evidence that such studies should be considered in individuals who appear to have isolated diaphragmatic defects.
Alterations in ZFPM2 underlie a small fraction of congenital diaphragmatic defects
Screening of the ZFPM2 gene in 52 patients with CDH and 10 patients with diaphragmatic eventrations failed to identify any clearly deleterious changes in the ZFPM2 gene. Similarly, Ackerman et al did not find deleterious ZFPM2 sequence changes in 29 additional postmortem samples from children with diaphragmatic defects.14 However, Bleyl et al screened 96 CDH patients—53 with isolated CDH, 36 with CDH and additional anomalies, and seven with CDH and known chromosomal anomalies—and identified a rare amino acid change, M703L, in a patient with isolated CDH that is predicted to be possibly damaging by PolyPhen and deleterious by SNPs3D (supplemental table 3).24 These data suggest that severe, deleterious alterations in ZFPM2 account for only a small portion of diaphragmatic defects. It is unclear, however, whether individual changes, like M703L, increase the risk for CDH and, if so, to what degree.
Delineation of new minimal deleted region for CDH on 1q41q42
Chromosome 1q41q42 has been shown to be recurrently deleted in individuals with CDH. The 1q41q42 minimal deleted region for CDH was previously defined by the maximal region of overlap between a patient reported by Kantarci et al and a patient described by Van Hove et al who were subsequently referred to as patient 7 and patient 4, respectively, by Shaffer et al.6 15 27 This interval spanned ∼6.3 Mb (220 120 384–226 397 002 hg19) and contained 48 RefSeq genes.
The maximal proximal and distal breakpoints of our patient 4's deletion are located inside the CDH region defined by Shaffer et al.15 These breakpoints can be used to define a new minimal deleted region for CDH on chromosome 1q41q42 which spans ∼2.2 Mb (223 073 839–225 318 623 hg19) and contains only 15 RefSeq genes (figure 2).
This refined minimal deleted region for CDH does not contain the H2.0-like homeobox (HLX) gene which has been considered a candidate gene for CDH based on its expression in the murine diaphragm and the diaphragmatic defects identified in Hlx−/− mice.28–30 Although it is possible that haploinsufficiency of HLX can contribute to the formation of diaphragmatic defects in humans, its exclusion from the minimal deleted region for CDH suggests that alterations in HLX function are not required for the development of CDH in association with 1q41q42 deletions.
The dispatched homologue 1 (DISP1) gene has also been considered a candidate gene for CDH and is located within the new minimal deleted interval for CDH. DISP1 is a membrane spanning protein that plays an essential role in SHH signalling.31 The potential role of DISP1 in the formation of CDH is supported by evidence that murine Disp1 is expressed in the pleuroperitoneal fold (PPF), a structure that is often considered to be the primordial mouse diaphragm and the identification of a somatic Ala1471Gly mutation—predicted to be benign by PolyPhen but ‘not tolerated’ by SIFT (http://blocks.fhcrc.org/sift/SIFT.html)—in a patient with CDH and other anomalies.32 However, abnormalities in other SHH signalling proteins have yet to be clearly shown to cause CDH. Truncating mutations (W475X and Y734X) in DISP1 have been found in two individuals with microforms of holoprosencephaly and their unaffected mothers—consistent with DISP1's role in SHH signalling—but not in individuals with diaphragmatic defects.32 33 This suggests that consideration should be given to the possible role of other 1q41q42 genes in the development of CDH. The tumour protein p53 binding protein, 2 (TP53BP2) gene could play a role in CDH development by modulating apoptosis and cell proliferation through interactions with other regulatory molecules and has been shown to play a critical role in heart and CNS development in mice.34 35 The WD repeat domain 26 (WDR26) gene is another candidate gene that is widely expressed and could play a role in the development of CDH by regulating cell signalling pathways and modulating cell proliferation.36 37
CDH is recurrently seen in 16p11.2 microdeletion syndrome
Recurrent 16p11.2 microdeletions result in a constellation of defects including developmental delay, cognitive impairment, behavioural problems, seizures, and congenital anomalies.17 Both patients 5 and 6 have de novo deletions of this region, suggesting that CDH should be added to the list of congenital anomalies associated with this microdeletion syndrome.
The T-box 6 gene (TBX6) is located on 16p11.2 and encodes a transcription factor that has been shown to play a critical role in important developmental processes including paraxial mesoderm differentiation and left–right patterning.38 39 As a result, TBX6 has been hypothesised to contribute to many of the congenital anomalies associated with 16p11.2.17 It is possible that haploinsufficiency of TBX6 also contributes to the development of CDH.
Evidence of CDH related genes on distal chromosome 11q and proximal chromosome 13q
Patient 7 carried an extra derivative chromosome 13 making her trisomic for a portion of chromosome 13 extending to 13q12.3 and a portion of chromosome 11 extending from 11q23 to the telomere—47,XX,+der(13)t(11;13)(q23.2;q12.3). A similar chromosomal complement 47,XY,+der(13)t(11;13)(q21;q14) was seen in a boy with CDH described by Park et al, suggesting that upregulation of genes on distal 11q and/or proximal 13q may play a role in the development of CDH.40 In support of this hypothesis, duplications of 11q have been recurrently seen in patients with CDH with many cases being associated with an unbalanced 3:1 meiotic segregation of the common t(11;22) translocation—resulting in a 47,XX or XY,+der(22)t(11;22)(q23.3;q11.2) chromosomal complement.13 41 Multiple cases of CDH associated with trisomy 13 have also been described providing support for the existence of one or more CDH related genes on chromosome 13.13
Role of FZD2 and Wnt signalling in the development of CDH and defects associated with pentalogy of Cantrell
The cardiac, diaphragmatic and anterior wall defects seen in patient 8 are similar to those described in pentalogy of Cantrell—a constellation of defects described by Cantrell et al that includes a supraumbilical omphalocele, a lower sternal cleft, a defect in the central tendon of the diaphragm, a defect in the pericardium, and an intracardiac anomaly.16 A de novo deletion of FZD2 was identified in patient 8, but similar deletions have been reported in normal individuals from Ibadan, Nigeria.20 Although this suggests that haploinsufficiency of FZD2 is unlikely to be the sole cause of this patient's phenotype, several lines of evidence suggest that decreased FZD2 expression may have contributed to this child's phenotype.
Frizzled genes encode a family of seven transmembrane domain proteins that act as receptors for Wnt signalling proteins.42 FZD2 is expressed in a variety of organs during development (supplemental table 1) and its protein product has been shown to interact with WNT3A and WNT5A and to signal through both the canonical (Wnt/β-catenin) and non-canonical (Wnt/Ca++ and cyclic GMP) Wnt pathways.43 In chick embryos, Wnt3a and Wnt5a modulate the migration of mesodermal precursors and Wnt3a has been shown to control the movement patterns of cardiac progenitor cells.44 45 Wnt signalling has also been shown to play a role in the development of CDH and omphalocele in humans. X linked dominant mutations in PORCN, a gene that encodes a protein that modifies Wnt proteins—including WNT3A—for membrane targeting and secretion, have been shown to cause CDH and omphalocele as part of focal dermal hypoplasia (Goltz syndrome; OMIM #305600).46 47
It is possible that deletion of FZD2, in combination with other genetic and/or environmental factors, caused dysregulation of these Wnt signalling pathways, leading to the development of the cardiac defects, CDH and omphalocele seen in patient 8. The potential role of frizzled signalling in developmental processes that require directional tissue movements followed by tissue fusion is supported by the work of Yu et al who recently showed that mouse frizzled 1 (Fzd1) and frizzled 2 (Fzd2) genes play an essential and partially redundant role in the correct positioning of the cardiac outflow tract and closure of the palate and ventricular septum.48 Genetic changes that disrupt these processes may ultimately be found to contribute to other cases of partial or complete pentalogy of Cantrell.
Genomic changes of unknown significance
The majority of the rare genomic changes catalogued in table 2 are not located within genomic regions that have been shown to be recurrently deleted or duplicated in individuals with CDH and do not affect genes/pathways that have been clearly implicated in the development of diaphragmatic defects. Although the significance of these changes is presently unknown, it is likely that future studies, in humans and animal models, will help to clarify whether these changes increase the risk of developing diaphragm defects.
Acknowledgments
The authors would like to thank the families who participated in this study.
References
Footnotes
Funding This work was supported by National Institutes of Health grants T32-GM007330-33S1 and F30-HL099469-01 (MJW), and KO8-HD050583 and R01-HD065667 (DAS). DV was supported by a Sophia-ErasmusMC grant SSWO 551. This work was also supported by Award Number P30HD024064 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development or the National Institutes of Health. These funding sources had no direct involvement in the study design, the collection, analysis and interpretation of the data, the writing of this report, and the decision to submit this manuscript for publication.
Competing interests None declared.
Ethics approval This study was conducted with the approval of the Institutional Review Boards of Baylor College of Medicine, Houston, Texas, USA and Erasmus Medical Center, Rotterdam, the Netherlands.
Provenance and peer review Not commissioned; externally peer reviewed.