Article Text

Abstract
Background X linked intellectual disability (XLID) is common, with an estimated prevalence of 1/1000. The expanded use of array comparative genomic hybridisation (CGH) has led to the identification of several XLID-associated copy-number variants.
Methods Array CGH analysis was performed using chromosomal microarray with ∼105 000 oligonucleotides covering the entire genome. Confirmatory fluorescence in situ hybridisation analyses were subsequently performed. Chromosome X-inactivation (XCI) was assessed using methylation-sensitive restriction enzyme digestion followed by PCR amplification.
Results A novel ∼0.5 Mb duplication in Xq28 was identified in four cognitively impaired males who share behavioural abnormalities (hyperactivity and aggressiveness) and characteristic facial features (high forehead, upper eyelid fullness, broad nasal bridge and thick lower lip). These duplications were inherited from mothers with skewed XCI and are mediated by nonallelic homologous recombination between the low-copy repeat regions int22h-1 and int22h-2, which, in addition to int22h-3, are also responsible for inversions disrupting the factor VIII gene in haemophilia A. In addition, we have identified a reciprocal deletion in a girl and her mother, both of whom exhibit normal cognition and completely skewed XCI. The mother also had two spontaneous abortions.
Conclusions The phenotypic similarities among subjects with int22h-1/int22h-2-mediated Xq28 duplications suggest that such duplications are responsible for a novel XLID syndrome. The reciprocal deletion may not be associated with a clinical phenotype in carrier females due to skewed XCI, but may be lethal for males in utero. Advancements in array CGH technology have enabled the identification of such small, clinically relevant copy-number variants.
- Chromosome Xq28 duplications
- chromosome Xq28 deletions
- X linked intellectual disability (XLID)
- factor VIII (F8) gene
- array comparative genomic hybridisation (array CGH)
- clinical genetics
- metabolic disorders
- cytogenetics
- getting research into practice
- cystic fibrosis
- copy-number
- diagnostics tests
- genetic screening/counselling
- genetics
- molecular genetics
- clinical genetics
- metabolic disorders
- cytogenetics
- getting research into practice
- copy-number
- diagnostics tests
- genetic screening/counselling, genetics
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Chromosome Xq28 duplications
- chromosome Xq28 deletions
- X linked intellectual disability (XLID)
- factor VIII (F8) gene
- array comparative genomic hybridisation (array CGH)
- clinical genetics
- metabolic disorders
- cytogenetics
- getting research into practice
- cystic fibrosis
- copy-number
- diagnostics tests
- genetic screening/counselling
- genetics
- molecular genetics
- clinical genetics
- metabolic disorders
- cytogenetics
- getting research into practice
- copy-number
- diagnostics tests
- genetic screening/counselling, genetics
Introduction
The prevalence of intellectual disability (ID) in developed countries is estimated to be around 2%–3%, and approximately one-third of ID cases are thought to have a genetic aetiology. Of these genetic causes, about one-quarter may be attributed to mutations on the X chromosome resulting in X linked intellectual disability (XLID), which has an estimated prevalence of 1/1000 males.1–4 To date, more than 200 different XLID disorders have been described, including around 150 syndromic forms, with over 80 XLID-associated genes having been identified (http://xlmr.interfree.it).
The expanded use of high-resolution genome analysis by array comparative genomic hybridisation (CGH) has led to the identification of several new microdeletion and microduplication syndromes.5–7 The application of arrays in XLID studies has resulted in the identification of new XLID-associated genes and copy-number variants (CNVs). For example, Froyen and colleagues screened a cohort of 108 subjects with ID by X-chromosome array CGH and identified X-chromosome CNVs in 14 subjects (13%).8 Duplications on the X chromosome correlated with ID more often than expected, suggesting a causal link between increased gene dosage and the disruption of normal cognitive development.3 The most common XLID-associated chromosomal aberrations are duplications of Xq28 comprising the MECP2 gene, which have been reported in more than 100 cognitively impaired individuals with characteristic facial features, hypotonia, seizures, speech delay and recurrent infections.9–12 Other known X-chromosome CNVs associated with XLID include deletions of Xp11.23, Xp11.3, Xp11.4 and Xp228 13–15 and duplications of Xp11.22, Xp11.22p11.23, Xp11.23, Xp11.4p21.3, Xp22.2 and Xq22.3.8 16–21
Cytogenetically visible duplications at Xq are rare, with only around 40 reported cases.22 23 The most frequent are distal Xq duplications involving Xq26q28, which have been reported in patients with severe ID, hypotonia, genital malformation and characteristic facial features.24–27 Less common are interstitial duplications at Xq21q24 that have been reported in patients with developmental delay, severe feeding difficulties, growth retardation and digital anomalies.22 23 Most Xq duplications observed in males are inherited from a mother exhibiting a normal or near-normal phenotype. Less frequently, Xq duplications are found in females with ID.23
We report four cognitively impaired males from three unrelated families with a novel ∼0.5 Mb duplication in Xq28 located telomeric to the MECP2 gene region and mediated by non-allelic homologous recombination between the low-copy repeat (LCR) regions int22h-1 and int22h-2, which are known to be involved in factor VIII (F8) gene inversions in subjects with severe haemophilia A. In addition, we report a reciprocal deletion in a mother and daughter with normal cognition. Herein, we describe the clinical phenotype of these subjects as well as the genomic structure of the deleted/duplicated region. In addition, we discuss the genes in this region and the potential effect of these rearrangements on the F8 gene.
Methods
Array CGH
Array CGH analysis was performed at the Medical Genetics Laboratories at Baylor College of Medicine. Informed consents were obtained as approved by the Institutional Review Board of Baylor College of Medicine. DNA was extracted from whole blood using the Puregene DNA extraction kit (Gentra, Minneapolis, Minnesota, USA) according to the manufacturer's instructions. The oligonucleotide-based chromosomal microarray version 7 (CMA V7 OLIGO) comprises approximately 105 000 oligonucleotides, which cover the entire genome at an average resolution of 30 kb with increased coverage at known disease loci. The array also includes six regions of known polymorphic variants.28 The procedures for DNA digestion, labelling and hybridisation were performed according to the manufacturer's instructions with some modifications. The slides were scanned into image files using a GenePix Model 4000B microarray scanner (Molecular Devices, Sunnyvale, California, USA) or an Agilent G2565 laser scanner, after which the image files were quantitated using Agilent Feature extraction software (version 9.0). Text file outputs from the quantitation analysis were imported to an in-house analysis package for copy-number analysis.29
Fluorescence in situ hybridisation analyses
Confirmatory fluorescence in situ hybridisation (FISH) analyses with bacterial artificial chromosome clones were performed on peripheral blood lymphocytes using standard procedures following the detection of copy-number changes via array CGH.30
Chromosome X-inactivation studies
Chromosome X-inactivation (XCI) studies were performed at the AR (androgen receptor)31 and FMR1 (fragile X mental retardation 1) loci.32 One hundred nanograms of genomic DNA from peripheral leucocytes of each female were digested with and without the methylation-sensitive restriction enzyme HpaII (New England Biolabs, Ipswich, Massachusetts, USA). DNA samples from male individuals were analysed without HpaII digestion. Five nanograms of DNA from each sample were subjected to PCR amplification with primers flanking the AR (CGA)n repeat region (5′ACCAGGTAGCCTGTGGGGCCTCTACGATGGGC3′ forward and 5′CCAGAGCGTGCGCGAAGTGATCCAGAACCCGG3′ reverse) and the FMR1 (CGG)n repeat region as previously described.31 32 PCR products were separated on an ABI 3770 Analyser and analysed with GeneMapper software. The XCI ratio was calculated as previously described.33 Inactivation ratios greater than 80:20 were designated as skewed XCI, whereas ratios greater than 95:5 were considered extremely skewed XCI.
Bioinformatics and in silico sequence analysis
Genomic sequence data based on the oligonucleotide coordinates from array CGH analysis were downloaded from the UCSC genome browser (Build 36, UCSC genome browser, March 2006). Regional assemblies were aligned using NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) in order to identify the LCR regions.
Results
Clinical description
Family 1 includes two brothers and their mother. The older brother is an 11-year-old who was born at term with a birth weight of 3.7 kg (50th–75th percentile) and a length of 56 cm (just above the 97th percentile). The pregnancy was complicated by gestational diabetes and cigarette smoking. During infancy, he experienced three episodes of pneumonia, all of which required hospitalisations. He was noted to be developmentally delayed beginning in early childhood. He started talking at the age of 6 years, started walking at 3 years and was toilet trained at 7 years. He is enrolled in special education classes and undergoes physical, occupational and speech therapy. At the time of this report, he was able to recognise letters and numbers and could write his name. However, he cannot read or solve simple mathematical problems. He exhibits autistic behaviours, including minimal eye contact and rocking, and was diagnosed as having Asperger syndrome. He also exhibits abnormally aggressive behaviours, including self-biting and hitting others. In addition, he presented with toe-walking, which required heel-cord lengthening, recurrent nosebleeds requiring multiple cauterisations, recurrent ear infections requiring ear tube placement, reactive airway disease and difficulty sleeping. Previous evaluations, including brain MRI, fragile-X DNA testing, and urine amino and organic acids, were all normal. Physical examination revealed a weight and height 3 SD above the mean and a head circumference at the 90th percentile. Distinctive facial features were also noted (table 1, figure 1A).
Clinical features, factor VIII levels and chromosome X-inactivation results for subjects with int22h-1/int22h-2-mediated Xq28 duplications and deletions

Facial features in individuals with int22h-1/int22h-2-mediated Xq28 rearrangements. (A) Facial features in the older brother in family 1, including high forehead, long face, upper eyelid fullness, open mouth and thick lower lip. Narrow, high arched palate, teeth crowding and overbite are not shown. (B) Facial features in the younger brother in family 1, including high forehead, upper eyelid fullness, broad nasal bridge, sparse eyebrows and thick lower lip. Small ears with simple helices and tongue tie with lingual frenulum are not shown. (C) Facial features in the mother in family 1, including high forehead, broad nasal bridge, sparse eyebrows and thick lower lip. (D) Facial features in the proband in family 2, including high forehead, upper eyelid fullness, deep-seated eyes, broad nasal bridge, thick lower lip and retrognathia. (E) Facial features in the proband in family 3, including high forehead, upper eyelid fullness, deep-seated eyes, broad nasal bridge and retrognathia. (F) Facial features in the proband in family 4, including high forehead, with no other dysmorphic features.
His younger brother is a 3-year-old born at term with a birth weight and length at about the 50th percentile. This pregnancy was also complicated by gestational diabetes and cigarette smoking. During the neonatal period, he was found to have a murmur, and an echocardiogram showed patent foramen ovale and patent ductus arteriosus, both of which had closed spontaneously prior to a subsequent echocardiogram. Similar to his older brother, he exhibited delays in attaining his developmental milestones. He started walking at 18 months of age and talking at 2 years. At the age of 3 years, he is only able to talk in simple two-word sentences. He receives physical and speech therapy. In addition, he has bilateral metatarsus adductus, requiring braces, and recurrent ear infections, which required ear tube placement. He also failed the newborn hearing screen and two subsequent hearing tests, and is being evaluated for a hearing aid. Physical examination revealed normal growth parameters and some distinctive facial features (table 1, figure 1B).
They have two maternal half-sisters, a 5-year-old enrolled in special education and a 14-year-old honour student, as well as two healthy 3- and 5-year-old paternal half-brothers. The parents are non-consanguineous. The mother is 32 years old and was reported to have learning difficulties. She required special education and did not continue schooling beyond the 11th grade. She was noted to have some similar facial features to her sons (table 1, figure 1C). The father has been healthy and did not report learning difficulties. The family history also revealed maternal uncles who required special education (figure 2).

Pedigrees for the three reported families with Xq28 duplications and the fourth family with the reciprocal Xq28 deletion.
Family 2 includes a 3-year-old boy evaluated for developmental delay and microcephaly, and his mother. He was born to a 16-year-old mother who reported usage of birth control pills for the first few months of pregnancy and had premature contractions at 6 months of pregnancy, which required hospitalisation. He was born at term with a birth weight around the 50th percentile and stayed in the neonatal intensive care unit because of hypoglycemia and jaundice. The mother had difficulty bonding with the baby, and he was therefore discharged with a foster family and was subsequently adopted. During the first few months of life, he was noted to have microcephaly. A skull x-ray was performed at the age of 9 months and showed no evidence of craniosynostosis. His medical history is significant for recurrent ear infections and a hospitalisation at the age of 7 months for pneumonia. Developmentally, he showed mild global delay, including rolling over at the age of 8 months, followed by cooing and sitting without support at 9 months. An assessment at 12 months indicated a developmental level of 9 months. Therefore, physical and speech therapy was initiated. By the age of 3 years, hyperactivity and aggression were noted, and physical examination revealed a head circumference within the third–fifth percentiles with height and weight around the 50th percentiles. He was also noted to have some distinctive facial features (table 1, figure 1D). He has a healthy 4-year-old maternal half-brother. No medical history was available for the biological father. His mother, who was reported to have learning difficulties, has a full brother and maternal half-brother, both of whom are intellectually disabled (figure 2).
Family 3 includes a boy and his mother. This 15-year-old male was born at term weighing 4.7 kg with a length of 55.9 cm, both of which are above the 95th percentiles. He was noted to be developmentally delayed during early childhood and was subsequently enrolled in special education as well as physical, occupational and speech therapy. Cognitive assessment at the age of 14 years revealed low ability across all cognitive domains with a full-scale IQ of 57. In addition, he has motor tics manifesting as head and neck twitching, has anxiety and was diagnosed as having attention deficit hyperactivity disorder. His medical history is significant for asthma, allergic rhinitis and recurrent ear infections requiring tube placement and adenotonsillectomy at the age of 5 years. For the past several years, he has been having recurrent episodes of erythema and oedema in his distal upper and lower extremities, joint pains, headaches and progressive muscle weakness in the forms of decreased exercise capacity and general slowness. Brain and spinal MRIs were normal, and a rheumatologic evaluation consisting of antiphospholipid and anticardiolipin antibodies was negative. He also presented with recurrent nosebleeds requiring repeated cauterisation, easy bruising and delayed clotting following skin laceration. His weight is 84 kg (>95th percentile), his height is 172.7 cm (75th–90th percentiles), and his head circumference is 56.7 cm (75th–90th percentiles). Additional findings on physical examination include lower extremity oedema, mild proximal muscle weakness and hypotonia with normal muscle bulk and reflexes, flat feet and distinctive facial features (table 1, figure 1E). As a result of his bleeding tendency, the factor VIII level was tested and found to be 24%, which is consistent with mild haemophilia A (table 1). A subsequent factor VIII inhibitor assay was negative, whereas VWF antigen and activity were within normal ranges. As part of factor VIII deficiency evaluation, F8 gene inversion analysis was performed using the method described by Liu and colleagues34 and was reported as positive for an F8 inversion. However, F8 gene inversions are associated with severe haemophilia A, which is inconsistent with the mild phenotype observed in this patient. Consequently, we realised that these results were misinterpreted for this patient as a result of the inherent limitations of F8 gene inversion analysis in the presence of an Xq28 duplication. This inversion analysis is performed by multiple PCR using four primers to differentiate between the normal F8 locus, inversions and carrier females (figure 3A–C). Although this patient does not harbour an F8 inversion, the results were misinterpreted as positive for an F8 inversion due to the presence of the Xq28 duplication (figure 3D). Subsequent sequencing of the F8 gene revealed hemizygosity for the point mutation c.6089G>A (p.S2030N), which has been associated with mild haemophilia A (http://hadb.org.uk/).35 The identification of this mutation explains the patient's mild haemophilic phenotype. An additional investigation included a karyotype, fragile X DNA testing, CPK level assay and thyroid function tests, all of which were normal. He has a healthy brother and maternal half-sister. His 41-year-old mother was reported to have learning difficulties, obesity, depression and hypothyroidism. The father is 42 years old and has hypothyroidism (figure 2).

Schematic representation of the Xq28 region demonstrating the F8 gene using arrowheads and int22h-1, 2 and 3 as black boxes with white triangles indicating their relative orientations.36 40 Panel (A) represents the normal genome, whereas panels (B–D) represent the results of different recombination events between int22h-1, 2 and 3, and their subsequent effects on the F8 inversion molecular testing described by Liu and colleagues.34 Two primers, P and Q, are located within the F8 gene at positions -1212 base pairs (bp) and +1334 bp flanking int22h1. Two primers, A and B, are located at -167 bp and +118 bp flanking int22h2 and int22h3. (A) In a normal male, segments PQ (12 kb) and AB (10 kb) are produced. (B) The int22h2/ int22h3-mediated inversion in males with haemophilia results in production of PB (11 kb) and QA segments (11 kb), along with the AB segment (10 kb) from the non-recombined int22h-2. (C) Females heterozygous for the inversion (carriers) produce PB (11 kb), QA (11 kb) and AB (10 kb) segments from the inverted allele, and PQ (12 kb) and AB (10 kb) from the normal allele. (D) Diagrammatic demonstration of non-allelic homologous recombination between int22h-1 and int22h-2, resulting in an int22h-1/int22h-2-mediated Xq28 duplication, which will result in formation of the AQ segment (11 kb) in addition to PQ (12 kb) and AB (10 kb), resulting in a pattern similar to that of a female heterozygous for the inversion.
Family 4 includes a girl and her mother. The 6-year-old girl was born at term after an uncomplicated pregnancy. She gained her developmental milestones appropriately during early childhood, but concerns have arisen regarding certain behaviours, including hyperactivity, inattentiveness, impulsivity, stereotypic movements (eg, running in circles and arm-flapping) and sensory-seeking behaviours (eg, licking her fist and putting her fingers in her mouth). Neurodevelopmental evaluation at the age of 5 years revealed sensory integration difficulties, for which occupational therapy was recommended. Beginning at 3 years old, she was also observed to have staring episodes. Brain MRI and continuous EEG monitoring were normal, and it was concluded that these episodes are non-epileptic in nature and are likely due to inattention. Physical examination revealed normal growth parameters, clinodactyly, a café-au-lait spot on her back and a high forehead with no dysmorphic features (table 1, figure 1F). Previous investigation, including fragile X DNA testing and thyroid function tests, was normal. She has a 1-year-old healthy sister. Her mother is a 39-year-old who has been healthy with normal intelligence, but had two spontaneous abortions at 5 and 8 weeks' gestation. Her father is 44 years old with depression and elevated cholesterol (figure 2).
Array CGH and FISH analyses
CMA V7 OLIGO performed on the four probands of families 1–3 revealed ∼0.5 Mb Xq28 duplications (153.7–154.2 Mb, hg18), which were confirmed by FISH analysis in each proband. The mothers in families 1–3 were also found to carry the same duplications by FISH. Furthermore, the proband's brother in family 3 was found to have a normal CMA. The proband in family 4 was found to carry a reciprocal Xq28 deletion and an ∼1 Mb 15q25.3 duplication (83.4–84.4 Mb, hg18). FISH determined that her father carries the 15q25.3 duplication, while her mother carries the Xq28 deletion (figures 2 and 4).
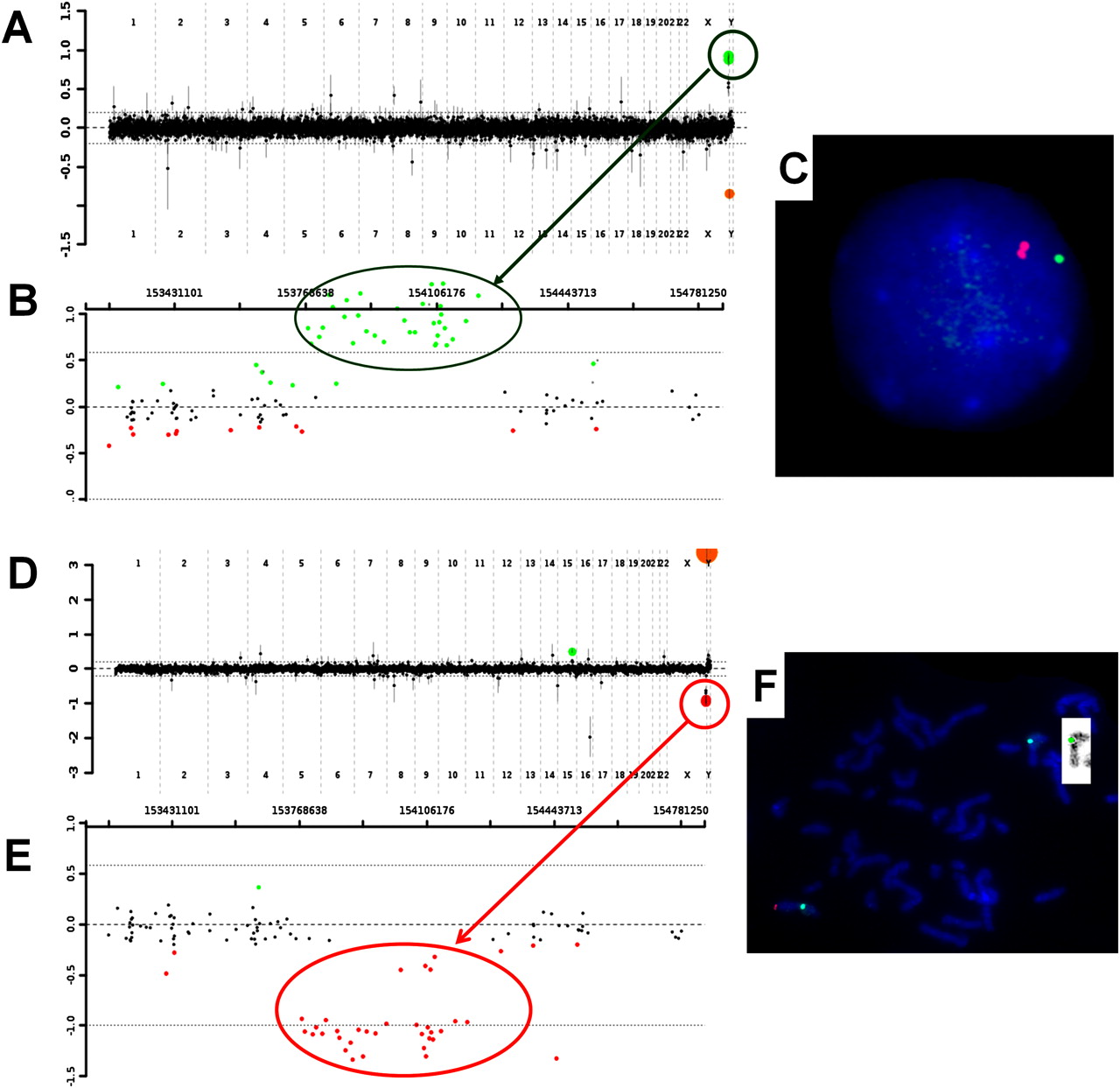
Panels (A), (B) and (C) demonstrate the chromosomal microarray and fluorescence in situ hybridisation (FISH) results for the proband of family 2. (A) Oligonucleotides are represented as green dots within the circle and indicate a gain of chromosome Xq28 material. (B) The Xq28 region, displaying individual oligonucleotides (green dots) that are displaced upward in the duplicated region. The proximal breakpoint is located between 153,759,353–153,774,852, and the distal breakpoint is located between 154,213,569–154,281,857. (C) FISH analyses showing the two bacterial artificial chromosome (BAC) clones, RP11-143H17 located within the duplicated region (red) and RP11-137H15 located proximal to the duplicated region and serving as a control (green). Panels (D), (E) and (F) demonstrate the chromosomal microarray and FISH results for the proband of family 3. (D) Oligonucleotides are represented as red dots within the circle and indicate a loss of chromosome Xq28 material. (E) The Xq28 region, displaying individual oligonucleotides (red dots) that are displaced downward in the deleted region. The proximal breakpoint is located between 153,759,353–153,774,852, and the distal breakpoint is located between 154,213,569–154,281,857. (F) FISH analyses showing the BAC clone RP11-143H17 (red) and the control probe targeting the centromere region of the X chromosome (green). The insert represents the reversed FISH image showing the deleted X chromosome.
XCI assay
XCI was assessed in the mothers of families 1, 2 and 3 at the AR and FMR1 loci whereby skewed XCI patterns were observed in all three mothers (table 1). Interestingly, the brothers in family 1 inherited the same AR allele (248) but opposite FMR1 alleles (281 vs 380). The AR 248 and the FMR1 281 alleles represent the preferentially active X chromosome in their mother, and it appears that both sons did inherit this same homologue but with one of them carrying the FMR1 380 allele most likely due to a recombination event. The finding that the preferentially active chromosome in the mother of family 1 is the same allele carried by her sons suggests that the mother's normal X chromosome is preferentially inactivated. Similarly, the assay at the AR and FMR1 loci in family 2 demonstrated that the preferentially active allele in the mother is the same allele carried by her son (AR 242 and FMR 278), which is consistent with the findings in family 1. That is, the normal X chromosome is preferentially inactivated, while the preferentially active X chromosome is that which carries the Xq28 duplication. Although the XCI assay at the AR locus was not informative for the mother of family 3, the assay at the FMR1 locus indicated preferential inactivation of the same allele carried by her son, suggesting that the X chromosome carrying the Xq28 duplication is preferentially inactivated in the mother. The daughter and mother in family 4 carrying the reciprocal Xq28 deletion showed 100% skewing XCI at the AR locus (table 1).
Xq28 genomic architecture
Using Blast2, we analysed the DNA sequence in the Xq28 region between 153.7 and 154.4 Mb, which harbours the duplication/deletion breakpoints, searching for alignments at least 2 kb in length with greater than 90% DNA sequence identity. The region contains three ∼9.5 kb LCR copies with 99.7% DNA sequence identity, one in intron 22 of the F8 gene and two located distally, which are consistent with the previously described int22h-1, int22h-2 and int22h-3 regions.36 The deletion and duplication breakpoints were localised within the directly oriented int22h-1 and int22h-2 regions. We identified two ∼50 kb LCR copies with 98.7% of the DNA sequence consistent with the previously described palindrome arms.36 In addition to these previously described regions, we also identified 10 LCR copies ∼5–6 kb in size and of 90%–94% DNA sequence identity (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the Xq28 region (153.7 and 154.4 Mb). The ∼0.5 Mb Xq28 duplicated/deleted region with minimum size of 0.439 Mb (153,774,852–154,213,569) and maximum size of 0.523 Mb (153,759,353–154,281,857) is represented as a red rectangle for each duplication and a green rectangle for the deletion. The F8 gene (153,717,258–153,904,192) is represented as a purple rectangle. The low-copy repeat (LCR) regions with DNA sequence identity >99% are also represented as red rectangles, the LCR regions with a DNA sequence identity of 98%–99% are represented as green rectangles, and the LCR regions with a DNA sequence identity of 90%–94% are represented as blue rectangles. The int22h-1 region is 9512 base pairs (bp) and spans 153,762,285–153,771,797; the int22h-2 region is 9553 bp in length and spans 154,259,350–154, 268,903, and the int22h-3 region is 9557 bp in length and spans 154,337,507–154,347,064. The proximal palindrome arm is 50374 bp in length and spans 154,219,197–154,269,571, and the distal palindrome arm is 50567 bp in length and spans 154,336,839-154,387,406.
Factor VIII assessments
Factor VIII levels were assayed in all probands. Only the proband in family 3 showed a reduced factor VIII level consistent with mild haemophilia A (table 1).
Discussion
In this report, we present the first evidence for the presence of int22h-1/int22h-2-mediated Xq28 rearrangements with an ∼0.5 Mb duplication in four cognitively impaired males who share behavioural abnormalities and characteristic facial features. The reciprocal deletion was detected in a cognitively normal girl and her mother.
The described rearrangement breakpoints were localised to the directly oriented 9.5 kb LCRs int22h-1 and int22h-2. The int22h-1 maps to intron 22 of the F8 gene, while int22h-2 and int22h-3 are located distally. Genomic inversions between int22h-1 and either int22h-2 or int22h-3 disrupt the F8 gene in nearly half of severe haemophilia A cases such that it was initially assumed that int22h-2 and int22h-3 were in opposite orientation to int22h-1.37 38 However, the publication of the sequence of the human X chromosome in 2005 revealed that int22h-1 and int22h-2 are, in fact, oriented in the same direction and are in opposite orientation to int22h-3.39 These same data also revealed that int22h-2 and int22h-3 are part of a palindrome, suggesting that recombination between the arms of this palindrome may establish a non-deleterious inversion polymorphism, which changes the relative positions and orientations of int22h-2 and int22h-3, thus permitting inversions involving int22h-1 and int22h-2. The low frequency of this polymorphic allele may explain the rarity of such inversions.40 41 Knowing that int22h-1 and int22h-2 are directly oriented, it has been predicted that recombination between int22h-1 and int22h-2 may also result in deletions and duplications. However, because of a lack of evidence, it has been suggested that they either are extremely rare or do not exist.36 39–42 Herein, due to advancements in high-resolution array CGH technology, we present evidence of such rearrangements in three families with int22h-1/int22h-2-mediated Xq28 duplication and in one family with the reciprocal deletion.
The int22h-1/int22h-2-mediated Xq28 duplications are approximately 0.5 Mb in size and are located telomeric to the MECP2 gene region. All four boys (families 1–3) with this duplication share characteristics that include cognitive impairment, behavioural problems (hyperactivity and aggressiveness), recurrent infections (pneumonia and ear infections) and characteristic facial features including high forehead, upper eyelid fullness, broad nasal bridge and thick lower lip (table 1, figure 1), suggesting that these duplications result in a distinct XLID syndrome. Furthermore, the absence of this duplication in the cognitively normal brother of the proband in family 3 lends additional support to this proposal. While the proband in family 3 also exhibits relative microcephaly, it is unclear whether the duplication is responsible.
The duplications in the probands in families 1–3 were all maternally inherited, and all three mothers were reported to have learning difficulties. Additionally, the mother of family 1 shared some of the facial characteristics present in the four male probands, including high forehead, broad nasal bridge and thick lower lip (table 1, figure 1). These observations suggest that int22h-1/int22h-2-mediated Xq28 duplications can be associated with intellectual impairment in carrier females.
XCI assessments are typically performed using primers at the AR locus (Xq12).31 This method can very accurately determine whether XCI is skewed or random. However, this method has limited utility in predicting whether the preferentially active or inactive X chromosome harbours the Xq28 duplication since the large distance between the AR locus and Xq28 (about 87 Mb) suggests a high likelihood of recombination. Therefore, we have also performed the assay at the FMR1 locus (Xq27), which is approximately 0.8 Mb from the duplicated region with a recombination frequency of less than 1%. Even so, the brothers in family 1 were found to have opposite alleles when they were tested at the FMR1 locus, suggesting a recombination event. We used both assays to predict the XCI status and to determine whether the X chromosome carrying the duplication preferentially remains active or is inactivated. Ultimately, the mothers in families 1–3 all demonstrated skewed XCI, and the results were concordant between the two methods (table 1). The XCI results also suggest that the normal X chromosomes are preferentially inactivated in the mothers of families 1 and 2. Most females with duplications involving the X chromosome demonstrate skewed XCI whereby the abnormal X chromosome is preferentially inactivated, which explains why the majority of carrier females do not manifest clinically. However, skewed XCI in favour of the X chromosome harbouring the duplication has been reported in females with Xp11.22p11.23 duplications who also exhibit a clinical phenotype attributed to the duplication.18 Although the mechanisms for such inactivation patterns are currently unknown, increased expression of a gene or genes in the duplicated region may provide unknown advantages to dividing cells.18 Thus, int22h-1/int22h-2-mediated Xq28 duplication may act in a similar manner to the Xp11.22p11.23 duplications such that the X chromosome carrying the duplication remains preferentially active. The fact that this abnormal X chromosome remains preferentially active in the mothers of families 1 and 2 may provide an explanation for the phenotypic effect of such duplications on those carrier females. However, the X chromosome carrying the duplication is found to be preferentially inactivated in the mother of family 3 who was also reported to have learning difficulties.
It is unclear why the X chromosomes that carry the duplication remain preferentially active in the mothers of families 1 and 2, but not in the mother of family 3. Similarly, it is also unknown why the mother of family 3 exhibits learning difficulties despite the abnormal X chromosome being preferentially inactivated. One possible explanation is a recombination event between the FMR1 locus and the Xq28 duplication region. The transposition of the duplication region to the opposite homologue would, in fact, yield results contrary to those of the XCI assessment performed at the FMR1 locus. This is because the duplication in the carrier mother would now be associated with FMR1 308 allele, which represents the active maternal chromosome. An alternative explanation involves the use of peripheral leucocytes for the XCI assays, such that the results exclusively reflect the XCI status of haematopoietic cells. It is possible that a distinct pattern of XCI occurs in the nervous system where the duplicated X chromosome may remain preferentially active. Such tissue-specific variation of XCI may provide a feasible explanation of the learning difficulties observed in the mother of family 3.33 42 Whereas our data are more consistent with the interpretation that the X chromosome carrying the Xq28 duplication remains preferentially active in the mothers of families 1 and 2, the observation of learning difficulties in the mother of family 3, whose Xq28 duplication is located on the inactive X chromosome, would argue against the significance of such genotype–phenotype correlation. Therefore, given the current inability to confirm any of the above theories, one cannot conclude decisively that the mothers' phenotypes are directly associated with an active abnormal X chromosome. Further studies of females with these duplications including assessment of X-inactivation status using additional markers within, and distal to, the duplication are needed to draw accurate conclusions as to the effect of such a duplication on the clinical phenotype and XCI in carrier females.
The mother and daughter in family 4 carry an int22h-1/int22h-2-mediated Xq28 deletion, although both are reported to have normal intelligence with no significant dysmorphic facial features. Since XCI showed a 100% skewed pattern in both individuals, these data suggest that the abnormal X chromosome is completely inactivated, such that these deletions are likely to have no phenotypic effect in females. However, it was noted that the mother had two spontaneous abortions. This observation coupled with the fact that this deletion has not been identified in males suggests that such deletions may be lethal for males in utero. Although family 4 includes the first cases of an int22h-1/int22h-2-mediated Xq28 deletion, one previous report indicated the presence of this deletion in a multi-generation family in which carrier females exhibited skewed XCI and experienced a higher than average rate of spontaneous abortion. The deletions were detected by FISH, and the breakpoints were roughly mapped using Southern analysis.43 44 The significant correlation between Xq28 deletion carrier status and the number of spontaneous abortions may support our hypothesis that this deletion is lethal for males in utero. Traditionally, chromosomal analysis has been recommended for females with recurrent spontaneous abortions, but int22h-1/int22h-2-mediated Xq28 deletions are beyond the resolution of standard karyotyping. We argue that these deletions can lead to recurrent spontaneous abortions in carrier females and therefore suggest considering array CGH as part of the investigation for multiple spontaneous abortions.
It has also been suggested that females carrying int22h-1/int22h-2-mediated Xq28 deletions should not exhibit clinical signs of haemophilia A as a result of the preferential inactivation of the X chromosome harbouring the F8 gene-inclusive deletion. In addition, it was proposed that males or females with int22h-1/int22h-2-mediated Xq28 duplications would not be at risk of haemophilia A because they maintain a normal copy of the F8 gene.41 Due to a lack of such cases, however, these hypotheses have not been tested until now. The patients reported here support the abovementioned suggestions. The daughter and mother in family 4 with the int22h-1/int22h-2-mediated Xq28 deletion did not report any bleeding tendency, and the daughter was found to have a normal factor VIII level. In addition, the probands in families 1 and 2 who harbour the reciprocal duplication did not present with significant bleeding tendencies or low factor VIII levels.
Interestingly, the proband in family 3 does demonstrate a bleeding tendency and was found to have a low factor VIII level consistent with mild haemophilia A. However, as previously indicated, the duplication should not result in a lower factor VIII level. Moreover, recurrent F8 gene inversions are generally associated with severe haemophilia A, which conflicts with the mild phenotype observed in this patient. It has been proposed that int22h-1/int22h-2-mediated Xq28 duplications may confound the molecular diagnosis of haemophilia A.45 Indeed, this was found to be true in the proband in family 3, whose F8 gene inversion analysis was performed using multiple PCR assay to identify inversions responsible for haemophilia A.34 Using this method, int22h-1/int22h-2-mediated Xq28 duplications result in a pattern indicative of a female inversion carrier (figure 3D). Therefore, this patient's results were falsely interpreted as positive for an F8 inversion. Considering that these duplications should not affect F8 gene expression, as well as the inherent limitations of F8 gene inversion analysis in the presence of such duplications, F8 gene sequencing was performed as a means of explaining the mildly haemophilic phenotype observed in the proband from family 3. And in fact, F8 sequencing revealed a hemizygous missense mutation that has been associated with mild haemophilia A. Hence, this is a clear example of how such duplications can confound the molecular diagnosis of haemophilia A via F8 inversion analysis. As a result, in cases of a positive inversion analysis with weak clinical correlation, we recommend the use of FISH or array CGH to rule out an int22h-1/int22h-2-mediated Xq28 duplication.
In addition to the F8 gene, which encodes coagulation factor VIII, the duplicated region contains nine other genes: H2AFB, F8A, FUNDC2, MTCP1NB, MTCP1, BRCC3, VBP1, RAB39B and CLIC2 (http://genome.ucsc.edu). H2AFB genes encode members of the histone H2A family.46 The F8A gene codes for the HAP40 protein, which co-purifies with the huntingtin protein, suggesting that HAP40 is likely to contribute to the function of normal huntingtin and is a candidate for involvement in the aberrant nuclear localisation of mutant huntingtin found in degenerating neurons in Huntington disease.47 The BRCC3 gene encodes a subunit of the BRCA1-BRCA2-containing complex, which is an E3 ubiquitin ligase. It is thought to be involved in the cellular response to ionising radiation and progression beyond the G2/M checkpoint. Therefore, it enhances cellular survival following DNA damage.48 The VBP1 gene encodes the Von Hippel–Lindau binding protein-1. Von Hippel–Lindau syndrome is associated with mutations in the VHL gene, which encodes the tumour-suppressor VHL. It was found that VHL acts as a molecular chaperone responsible for carrying Von Hippel–Lindau binding protein-1 from perinuclear granules to the nucleus or cytoplasm.49 Expression in mice during fetal stages was primarily limited to the central nervous system, retina and liver, whereas adult expression was more evenly distributed.50 The RAB39B gene encodes a member of the Rab family of proteins, which are small GTPases that are involved in vesicular trafficking.51 CLIC2 encodes the chloride intracellular channel 2 protein, which shares 60% sequence identity with the chloride intracellular channel 1 protein, a nuclear chloride channel.52 It is expressed in fetal liver and adult skeletal muscle and has been suggested to function as an intrinsic stabiliser of ryanodine receptors in skeletal muscles.53 Nevertheless, none of the aforementioned genes have been shown to be associated with XLID, although increased dosage of one or more of those genes may affect brain development, resulting in cognitive impairment.
In conclusion, int22h-1/int22h-2-mediated Xq28 duplications may represent a novel XLID syndrome resulting in cognitive impairment, behavioural abnormalities, recurrent infections and characteristic facial features in males. Carrier females may exhibit a clinical phenotype with skewed XCI. Such duplications do not affect factor VIII levels, but can result in errors in the molecular diagnosis of haemophilia A. Alternatively, the reciprocal deletion in females may not have phenotypic consequences as it appears to result in completely skewed XCI. However, these deletions may be lethal for males in utero, resulting in recurrent spontaneous abortions in carrier females.
References
Footnotes
Correction notice This article has been corrected since it was published Online First. The authors' competing interests have been added.
Competing interests Dr Sau Wai Cheung is the director of the Cytogenetics Laboratory, Department of Molecular and Human genetics, Baylor College of Medicine. The cytogenetic and molecular laboratories at Baylor College of Medicine offer extensive genetic laboratory testing including use of array CGH for genomic copy number analysis and derive revenue from this activity.
Ethics approval Institutional Review Board of Baylor College of Medicine.
Provenance and peer review Not commissioned; externally peer reviewed.