Article Text

Abstract
Background The cystic fibrosis (CF) basic defect, caused by dysfunction of the apical chloride channel CFTR in the gastrointestinal and respiratory tract epithelia, has not been employed so far to support the role of CF modifier genes.
Methods Patients were selected from 101 families with a total of 171 F508del-CFTR homozygous CF patients to identify CF modifying genes. A candidate gene based association study of 52 genes on 16 different chromosomes with a total of 182 genetic markers was performed. Differences in haplotype and/or diplotype distribution between case and reference CF subpopulations were analysed.
Results Variants at immunologically relevant genes were associated with the manifestation of the CF basic defect (0.01<Praw<0.0001 at IL1B, TLR9, TNFα, CD95, STAT3 and TNFR). The intragenic background of F508del-CFTR chromosomes determined disease severity and manifestation of the basic defect (Praw=0.0009). Allele distributions comparing transmitted and non-transmitted alleles were distorted at several loci unlinked to CFTR.
Conclusions The inherited capabilities of the innate and adaptive immune system determine the manifestation of the CF basic defect. Variants on F508del-CFTR chromosomes contribute to the observed patient-to-patient variability among F508del-CFTR homozygotes. A survivor effect, manifesting as a transmission disequilibrium at many loci, is consistent with the improvement of clinical care over the last decades, resulting in a depletion of risk alleles at modifier genes. Awareness of non-genetic factors such as improvement of patient care over time is crucial for the interpretation of CF modifier studies.
- Cystic fibrosis
- modifier genes
- association study
- affected sib pairs
- CF basic defect
- genetics
- immunology (including allergy)
- infectious diseases
- epidemiology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Cystic fibrosis
- modifier genes
- association study
- affected sib pairs
- CF basic defect
- genetics
- immunology (including allergy)
- infectious diseases
- epidemiology
Introduction
Cystic fibrosis (CF) is the most common severe, monogenic, autosomal recessively inherited disease, affecting 1 out of 3200 newborns in the Caucasian population.1 The manifestation of the multiorgan disease varies among individuals and even among siblings homozygous for the most frequent disease-causing lesion F508del-CFTR.2–4 Improvement of medical care throughout the last decades has increased the patient's life expectancy from less than 5 years in the 1940s to 37 years estimated for the current CF population,5 6 demonstrating convincingly that mainly non-genetic factors determine the disease outcome. Hallmarks of improved symptomatic therapy are the use of antibiotics and nutritional repletion in centres specialised for CF care, starting in the mid-1950s,5 and the development of specialised drugs such as novel pancreatic enzymes or recombinant DNAse since the late 1980s.5 Centre to centre variations in quality of care7 and the influence of socioeconomic status on the course of CF disease6 furthermore emphasise the influence of non-inherited factors on disease manifestation. Consequently, attempts to uncover clinically meaningful modifier genes require that non-genetic confounding factors are taken into account.
The basic defect in CF is defined as an impaired chloride conductance in epithelia that express CFTR, a chloride channel localised in the apical membrane of differentiated epithelial cells.1 Nasal potential difference measurement (NPD) and intestinal current measurement (ICM) allow the assessment of the CF basic defect in the two major affected organ systems in CF—the respiratory tract and gastrointestinal tract.8 9 Even though the manifestation of the basic defect on the cellular and epithelial level is less vulnerable to environmental influences than any clinical outcome parameter such as lung function, growth and stature, substantial variability of the basic defect phenotype among sib pairs with shared CFTR mutation genotype is observed.10 11 We hypothesise that CF genetic modifiers can be recognised through a genotype–phenotype association with the basic defect.
If the influence of modulating genes on the CF phenotype is at the core of interest, the power of the study to detect a modifying gene will increase, with the standardisation of the CFTR mutation genotype1 2 5 and environmental influences1 5–7 as the two major confounding variables of CF disease severity. While the high frequency of the F508del-CFTR allele among Caucasians1 2 allows the recruitment of a sufficient number of patients who are homozygous for the same CFTR disease-causing lesion,3 12 13 standardisation of the environment cannot be achieved for a human study population. Hence, the best choice are twin and sibling patient pairs to minimise many environmental effects as siblings will share their family environment, visit the same physician and be treated at the same CF centre. Monozygous twins, being genetically identical, and dizygous patient pairs, sharing half of their genome, can be compared to deduce the influence of inherited factors on the phenotype.14
The European Cystic Fibrosis Twin and Sibling Study Consortium3 has collected clinical data on more than 500 CF twin and sibling pairs. We have previously reported how informative patient pairs with extreme phenotypes can be selected from this cohort for candidate gene analysis3 and described CF modifier genes on six chromosomes.15–22 In this study, our objectives are: (1) to qualify the relative impact of the CFTR gene, environmental and other inherited factors on CF disease severity; (2) to identify genetic modifiers for the CF basic defect; and (3) to observe whether or not these are the same genes that modify CF disease severity. We have tested a total of 182 genetic markers targeting 52 candidate gene loci on 16 different chromosomes for their association with CF disease severity and the manifestation of the CFTR mediated basic defect, by genotyping a cohort composed of 101 families with a total of 171 F508del-CFTR homozygous patients. Among these are 12 genes studied as CF modifiers by others,12 13 23–26 26 additional genes which represent plausible modifier candidates such as direct interaction partners of CFTR, alternative ion channels and components of the innate immune system, and 14 candidate genes which were selected by transcriptome analysis comparing intestinal epithelial biopsies of controls and CF patients. To our knowledge, this is the only study on CF modifiers that combines the powerful approach to analyse affected sib pairs13 while at the same time exploiting the phenotypic contrast between informative extreme clinical phenotypes.12
Methods
Patients and phenotypes
This study reports on clinical data from 466 twin and sibling pairs and genetic data from 101 CF families, 85 of which are a subgroup of the twin and sibling study cohort. Enrolment of patient pairs into the association study was based on one of the following criteria: the twin or sibling pair exhibits an extreme clinical phenotype3 and/or the patient was characterised for the CF basic defect.10 11 Sixteen unrelated F508del-CFTR homozygotes from the CF clinic in Hannover were included who participated in an analysis of the basic defect and transcriptome in the intestine to define 14 candidate genes (van Barneveld et al, 2010, unpublished data).
We selected the 12% most informative pairs from the entire sample of 318 CF twin and sibling pairs by a ranking algorithm3 that relies on two clinical parameters most sensitive for course and prognosis of CF.27 28 These 37 dizygous F508del-CFTR homozygous patient pairs, enrolled due to their contrasting extreme clinical phenotype (Figure 1), were comparable with respect to their birth cohort.18

Assembly of patient subsets for genetic modelling and association study. Weight, height, and lung function data of at least one out of two siblings was received in 1995–1996 for 540 cystic fibrosis (CF) sib pairs from 158 CF clinics in 14 European countries. Pairs for whom clinical data was obtained for both sibs of a pair were included into genetic modelling (466 pairs with weight and height, 318 pairs with lung function).3 Subsamples of F508del homozygotes were selected for the association study based on the clinical phenotype or the basic defect phenotype which was measured by nasal potential difference measurement (NPD) and intestinal current measurement (ICM).10 11 In total, 171 patients from 101 families were genotyped. Two patients with CFTR mutation genotypes other than F508del/F508del were only included for the phenotype ‘DIDS-sensitive chloride conductance’ determined by ICM. The association study compared subsets of affected patient pairs in their manifestation of the clinical phenotype, and correspondingly only dizygous pairs were included. In case of case–control comparisons of the basic defect in subsamples of unrelated index cases, one twin per monozygous twin pair was added to the cohort. For the association study on ICM phenotypes, 16 families with F508del homozygous offspring recruited from the CF clinic in Hannover, Germany were included. Integers within this figure correspond to the number of independent families while all numbers in brackets refer to the number of individual patients. Relative proportions of samples and subsamples stratified for extreme phenotypes are displayed on the basis of the number of independent nuclear families—that is, of sib pairs instead of individual patients—in order to represent sample sizes based on their number of independent genetic contributions which define the effective number of chromosomes considered for the genetic association study. Integers on top of diagrams report the sample size as the sum of all subgroups. Integers adjacent to diagrams report sizes of subgroups which are visualised as slices. For subgroups consisting of 540, 466 and 318 pairs, the entire circle diameter corresponds to the sample size. For subgroups consisting of 46 and 55 families, the sample size is represented in proportion to the larger datasets by the circle diameter carrying the sample size label (white central circle with red rim on top of the diagram). Composition of subgroups stratified for manifestation of the basic defect is displayed on the basis of individual patients. The numbers displayed for the contrasting phenotypes analysed in the case–control scenarios show the effective sample size, taking into account that sib pairs who show a discordant basic defect phenotype can contribute to one out of two (NPD phenotypes) or one out of three (ICM phenotypes) subgroups only in the association study. In other words, subsets of unrelated patients were defined by an index case strategy whereby extreme basic defect phenotypes were selected for in pairs for whom discordance ICM or NPD was observed. As NPD measurements were obtained for two siblings of a pair in most cases, two subsets for each case–control test were evaluated to provide an internal control depending on which sibling was assigned at random to be the index case. Candidate genes were interrogated for their association with disease severity using the phenotypic contrast between concordant/mildly affected patient pairs, concordant/severely affected patient pairs, and discordant patient pairs. For this purpose, 37 dizygous pairs—representing the 12% most informative patient pairs of the entire sample—were selected based on a non-parametric ranking algorithm as described previously.3 NPD data were used firstly to enquire for an association with sodium transport via the amiloride sensitive proportion of the potential, using the upper and lower 30% of the entire sample to define extreme phenotypes, secondly to evaluate an association with residual chloride secretion activated by chloride-free gluconate solution and isoproterenol, and thirdly to analyse a contribution of the selected candidate genes on ATP stimulated chloride response. The basic defect assessed by ICM was evaluated using a set of patients devoid of residual chloride secretion as controls. The pleiotropic chloride channel inhibitor DIDS, to which CFTR is not sensitive at the chosen concentration, was used to discriminate CFTR mediated residual chloride secretion from chloride secretion mediated by alternative channels which are DIDS sensitive.10 11
CF basic defect phenotypes derived from intestinal current measurement (ICM) and nasal potential difference measurement (NPD) were obtained for 71 patient pairs.10 11 Briefly, secretagogues that activate or block ion channels, ion exchangers or components of the cellular signal transduction pathways were applied by superfusion of the surface of the lower nasal turbinate9 or to excised rectal suction biopsies mounted in a micro-Ussing chamber8 using a fixed sequence of defined pharmacologically active substances.
Based on the sib pairs' clinical phenotype and the individual's manifestation of the CF basic defect, subgroups were defined as cases and references for an association study (figure 1). These subgroups are by definition partially (clinical phenotype vs ICM) or nearly completely (ICM and NPD phenotypes) overlapping sets of patient pairs (see supplementary material for details).
This study was approved by the ethics committee of the Medizinische Hochschule Hannover. Written informed consent was obtained from all participants or their parental guides.
Candidate genes and genetic markers
Genotypes of two indels, 101 single nucleotide polymorphisms (SNPs) and 79 microsatellites were analysed for this study. Seventy-three markers were developed de novo for this study, building on raw genomic sequences containing microsatellite repeats within or near the targeted candidate gene to develop polyallelic markers (40 microsatellites) or on the public SNP database (33 diallelic markers). To compensate for the loss of power inflicted upon the study by the limited sample size of subgroups stratified for extreme clinical phenotypes (being infrequent by definition) or for basic defect phenotypes such as F508del-CFTR homozygotes who display CFTR mediated residual function (being infrequent due to the aetiology of the disease), care was taken to include only informative markers in the study: between one and 13 microsatellite motifs (median: four motifs) were tested per candidate gene locus to obtain one marker with a suitable polymorphism information content of 0.61–0.91 (mean 0.76) per targeted candidate gene. SNP markers were preselected based on HAPMAP data.29 Technical details on all markers and genotyping methods are provided in the supplementary material.
Genetic modelling
Genetic modelling was done with the LISREL8 software package.30 Briefly, covariance matrices from standard normal distributions derived from weight for height and forced expiratory volume in 1 s as percentage of predicted value (FEV1%pred) data were used to fit a model taking genetic effects as well as random and shared environmental effects into account. Applicability of the model was judged by the Aikaike information criterion.30
Association study
Genetic data for the association study was evaluated using the FAMHAP software package31 which allows family based analysis32 33 and accepts data evaluation in association studies on unrelated individuals as well as on affected sib pairs.31 All case–reference comparisons were carried out using 10.000 Monte-Carlo simulated datasets.31–33 Nuclear families were analysed by the transmission–disequilibrium test (TDT)34 extended to both nuclear families with more than one affected child and to multi-marker haplotypes.31–33 To allow a comparable assignment of weighted haplotype explanations, the entire genotyping data were provided as training set to FAMHAP for all case-reference comparisons.
All candidate genes were analysed for an underrepresentation of alleles among transmitted chromosomes (TDT) as well as by case–reference comparisons for their association with disease severity (two case–reference comparisons), ICM (two case–reference comparisons) or NPD (three case–reference comparisons). Assignment of case and reference status is outlined in figure 1 and detailed in the supplementary material. To confirm observed associations, we have employed within-dataset validation strategies using partial replicates as detailed in the supplementary material. Significance within the entire dataset of 182 markers was conservatively judged by Bonferroni correction for 200 independent markers (threshold Praw=3×10-4 for α=0.05) or 52 independent loci (threshold Praw=9×10-4 for α=0.05).
Results
Inherited, environmental and maternal factors influence CF disease manifestation
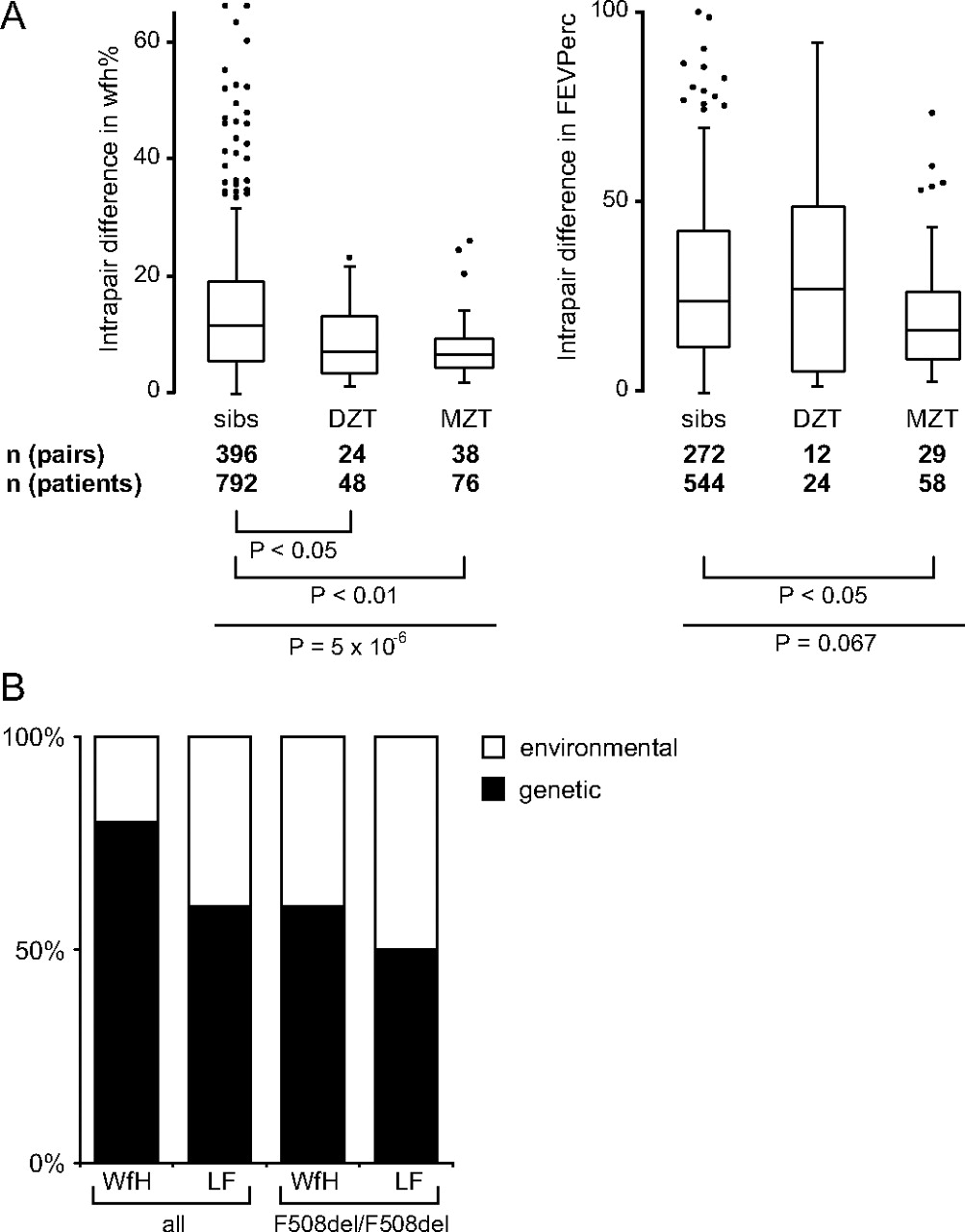
We have compared intrapair differences in weight expressed as percentage of predicted weight for height (wfh%) and CF population centiles for the lung function parameter FEV1% (FEVPerc) and conducted genetic modelling on 466 (wfh%) and 318 (FEV1%) patient pairs (figure 2). While monozygous twins were more concordant in their FEVPerc than either dizygous twins or sibs, all twins were more concordant than sibs in their wfh% irrespective of their zygosity status. This indicates that the shared pre- and early postnatal period which distinguishes twins from sibs is associated with lower intrapair differences in wfh% while concordance in lung function is dominated by genetic factors as only monozygous twins share their entire genetic information in contrast to dizygous twins or siblings who have half of their genetic information in common. The results of the genetic modelling analysis imply that inherited factors have a larger impact on the patient-to-patient variation in wfh% than for lung function (figure 2B). These data are in accordance with heritability estimates from other CF twin and sibling cohorts.4
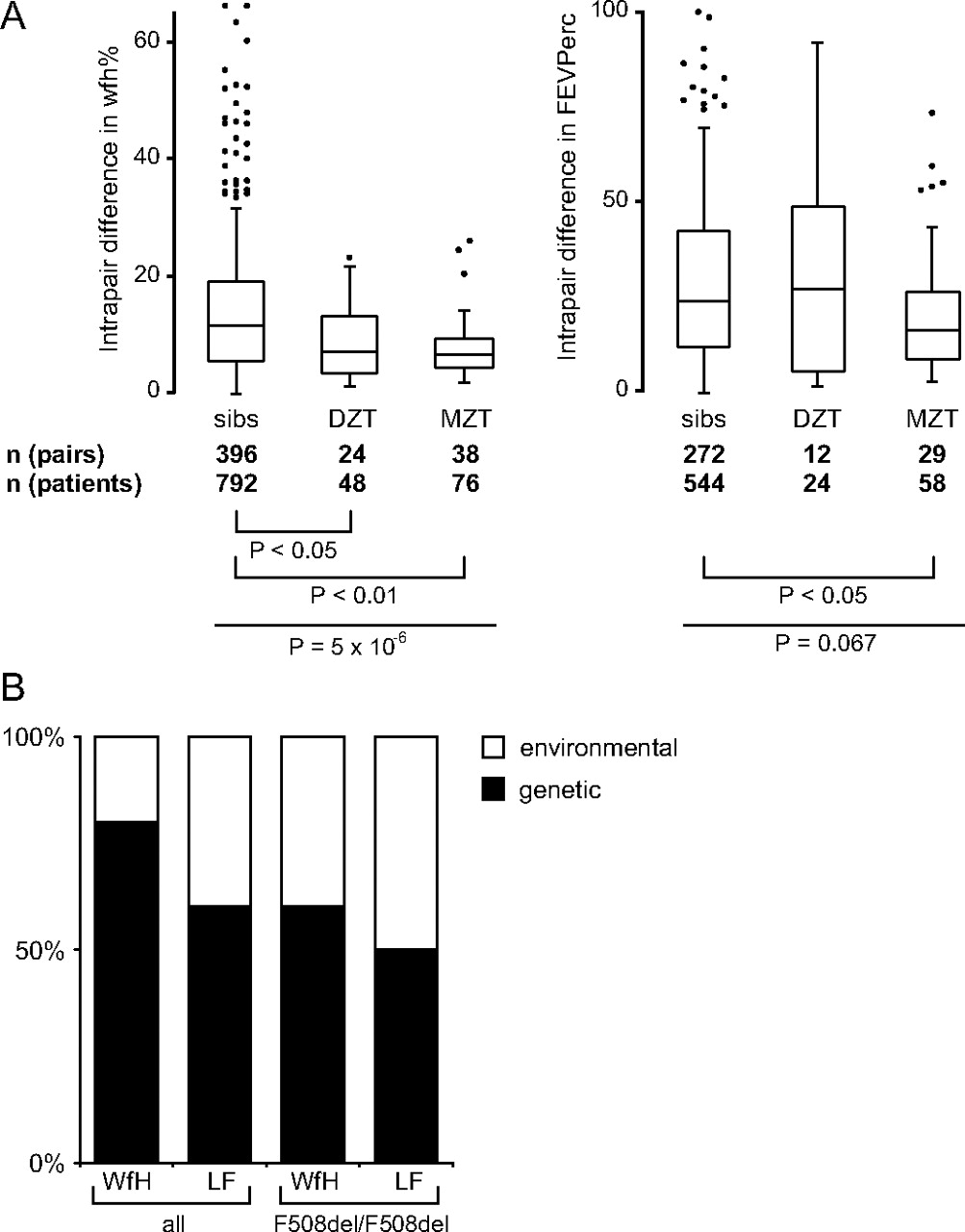
Relative impact of environmental and inherited factors on cystic fibrosis (CF). (A) Distribution of intrapair differences in weight for height % (wfh%, left) and the CF population centiles for FEV1%pred (FEVPerc, right) among sib pairs, dizygous twins (DZT) and monozygous twins (MZT). Overall comparisons were done by Kruskal-Wallis rank test (wfh%: P=5×10-6; FEVPerc: P=0.067) and individual subgroups were compared by Dunn rank test. Please note that both monozygous and dizygous twins are similarly concordant in wfh%, indicating that the shared in utero environment determines outcome in wfh%. In contrast, monozygous twins are more concordant that either dizygous twins or sibs in FEVPerc, implying that lung function is influenced by inherited factors. (B) Relative impact of inherited and environmental factors on disease manifestation as estimated by genetic modelling. Covariance matrices of weight as % of predicted weight for height (WFH) and forced expiratory volume in 1 s as % of predicted value (LF) were analysed by genetic modelling.30 All patient pairs and the subgroup of F508del-CFTR homozygotes were evaluated. Models were based on a linear combination of the four following factors: random environmental effects, shared environmental effects, additive and dominant genetic effects. Goodness-of-fit was judged by χ2 measure, whereby in case of two or more models with a similar goodness-of-fit the least complicated model, based on the fewest out of the four linearly combined factors, was accepted as suggested by the Akaike information criterion.30 Consensus diagrams displaying the impact of inherited (black bars) and environmental (white bars) factors are shown.
Association study
Candidate genes were interrogated for their association with disease severity using the phenotypic contrast between concordant/mildly affected patient pairs, concordant/severely affected patient pairs, and discordant patient pairs. For this purpose, 37 dizygous pairs—representing the 12% most informative patient pairs of the entire sample—were selected based on a non-parametric ranking algorithm as described previously.3 NPD data were used: first, to enquire for an association with sodium transport via the amiloride sensitive proportion of the potential, using the upper and lower 30% of the entire sample to define extreme phenotypes; secondly, to evaluate an association with residual chloride secretion activated by chloride-free gluconate solution and isoproterenol; and thirdly to analyse a contribution by the selected candidate genes on adenosine triphosphate (ATP) stimulated chloride response. Basic defect assessed by ICM was evaluated using a set of patients devoid of residual chloride secretion as controls. The pleiotropic chloride channel inhibitor DIDS, to which CFTR is not sensitive, was used to discriminate CFTR mediated residual chloride secretion from chloride secretion mediated by alternative channels which are DIDS sensitive.10 11 After correction for multiple testing, significance was retained at the following loci and for the following phenotypes (figure 3): 7q31.1/CFTR: P=0.0009 (NPD_Na+) and Praw=0.0008 (NPD_ATP); 7q34/D7Sat3: Praw=0.0005 (disease severity cis); 11q13/GSTP1 Praw=0.0006 (NPD_ATP); 12p13/TNFR Praw=0.0004 (NPD_Gl/Iso) and Praw=0.0003 (NPD_ATP); 12q13/KRT8 Praw=0.0004 (ICM_Res); 16p13.3/NHERF2 Praw=0.0001 (ICM_Res). We have previously reported an association with DIDS-sensitive residual chloride secretion in ICM at the CLCA-gene cluster15 and an association with disease severity for LEP,16 CD95,17 TNFR,18 SCNN1B,18 SCNN1G,18 at the CEACAM gene cluster,19 20 and at a paternally imprinted gene on 7q34.21 Association of these genes with other phenotypes are reported here for the first time.

Association of candidate genes with CF disease severity and basic defect. Candidate genes are listed according to their position in the human genome. Association to genes instead of results of individual markers are displayed if several linked markers were analysed per gene or gene cluster. For PRSS8, SCNN1B/SCNN1G, KRT8/KRT18, TNFR/SCNN1A, TLR4, the results of the microsatellite scan and the confirmation by low resolution single nucleotide polymorphism (SNP) typing are shown. Solid lines are used to indicate unlinked genes, adjacent loci within 10 Mb distance are separated by dotted lines and neighbouring genes are not separated by lines between gene names. All candidate genes were analysed for an underrepresentation of alleles among transmitted chromosomes (TDT) as well as by case–reference comparisons for their association with disease severity, intestinal current measurement (ICM) or nasal potential difference measurement (NPD): disease severity cis—case = concordant mildly affected patient pairs, reference = concordant severely affected patient pairs16–18; disease severity trans—case = discordant patient pairs, reference = concordant patient pairs16 17; ICM_Res—case = DIDS-insensitive (CFTR-mediated)10 11 residual chloride secretion, reference = no residual chloride secretion; ICM_DIDSRes—case-DIDS-sensitive (≠CFTR)10 11 residual chloride secretion, reference = no residual chloride secretion; NPD_Na+—case = low response to amiloride, reference = high response to amiloride; NPD_Gl/Iso—case = no response to gluconate and isoproterenol, reference = residual chloride secretion response to gluconate and isoproterenol; NPD_ATP—case = no response to ATP, reference = residual chloride secretion response to ATP. Solid dots correspond to raw P values for single chromosomes (marked H), chromosome pairs (marked D) or accumulation of rare variants (marked r). All displayed values are uncorrected Praw (see also supplemental table 8).
Informative microsatellite markers allow the assessment of candidate genes as CF modifiers
Most candidate gene association studies in CF rely on the typing of polymorphisms which are suspected or known to alter the performance of the gene or its protein product,12 23 a strategy that cannot be applied as long as functional variants are unknown to date. Building on our previous experience,18 22 we developed informative microsatellite markers to interrogate these genes (see supplementary material for details). Results of the association study are displayed in figure 3. To confirm initial findings by microsatellite markers, a low density SNP scan was done for PRSS8, KRT8/KRT18, TLR4 and IL1B, and previously obtained genotyping data18 was evaluated for basic defect phenotypes at SCNN1B/SCNN1G and TNFR/SCNN1A. Significance after correction for multiple testing was retained for the association of the member of the CFTR network NHERF2 with DIDS insensitive residual chloride secretion detected by ICM (Praw=0.0001, diplotype model; Praw=0.0007, haplotype model; Praw<10-5, accumulation of rare alleles among patients with residual function; figure 3 and figure 4). NHERF2 was interrogated with two microsatellite motifs located at a distance of 30 kb and 70 kb. Seven further genes are encoded on the segment between marker and targeted candidate gene. Even though none of these can be reasonably associated with CF pathophysiology, neighbours of the targeted candidate gene have to be considered; we observed an association of the response to gluconate and isoproterenol in NPD at the TNFR/SCNN1A locus (figure 3). Haplotype analysis of the 12p13 locus with four intragenic markers for each of the two neighbouring genes TNFR and SCNN1A revealed that the effect is mediated by the cytokine receptor TNFR and not by the α subunit of the amiloride sensitive epithelial sodium channel SCNN1A (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of allele distributions among patients with different basic defect phenotypes. Allele distributions are shown for the CFTR-locus (A–C), SCNN1A, encoding the α-subunit of the epithelial sodium channel ENaC and the neighbouring gene TNFRSF1A, encoding the 55 kDa receptor for TNFα (TNFR) (D, E) and the CFTR interaction partner NHERF2 (F). Pictograms represent the relative location of targeted candidate gene (black box) and genotyped markers (vertical lines). Markers are: XV-2c, the variant HUG16RS35 and J3.11 (A, B); XV-2c, KM19 5' and IVS17bTA36 (C); four markers in SCNN1A (nt7AG, SC3, SC4 and rs2228576 (D); four markers in TNFR (rs767455, D12S889, rs1800692 and rs1800693 (E); two microsatellites near NHERF2 (F). Alleles at IVS17bTA are calibrated in accordance with Morral et al.37 For other microsatellites, arbitrary repeat units were assigned with 10 corresponding to the most frequently observed allele. Single nucleotide polymorphisms (SNPs) typed by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) are named according to the presence (allele 2) or absence (allele 1) of the diagnostic restriction site. P values and corresponding allele distributions (A–B; D–F) or diplotypes distributions (C) are given for six case–control tests. Pbest is the best observed P value while Pglobal was corrected for multiple testing33 of all markers at the locus. Phenotypes are: NPD_Amil: NPD change to amiloride [mV]; NPD_ATP: NPD change to ATP; NPD_GI: NPD change to gluconate and isoproterenol [mV]; ICM no Res.: no residual response; ICM Res.: cAMP sensitive and DIDS insensitive residual chloride secretion. The four-marker haplotype at TNFR correlates with cystic fibrosis (CF) disease severity18 whereby allele 1-10-2-2 is associated with a mild CF phenotype (E) and is also overrepresented among patients exhibiting residual chloride secretion assessed by nasal potential difference measurement (NPD) (D).
Diversity of the F508del-CFTR genetic background determines the clinical variability of F508del-CFTR homozygotes
We have included intragenic and CFTR flanking markers in our association study. As the enrolled patients are F508del-CFTR homozygous, the major disease causing variant is identical for all study subjects. In contrast, these F508del-CFTR chromosomes differ at closely linked neighbouring SNPs.35–37 We tested in our association study whether these different F508del-CFTR alleles are asymmetrically distributed when comparing mildly and severely affected patients or subgroups stratified for the manifestation of the basic defect. We observed a significant association of the CFTR core haplotype to the response of the nasal epithelium to superfusion with amiloride (Praw=0.0009; figure 3 and figure 4) and ATP (Praw=0.0008; figure 3 and figure 4). Moderate association of CFTR was observed with CF disease severity and response to gluconate and isoproterenol in NPD (0.001<Praw<0.01; figure 3 and figure 4). These findings demonstrate that the different F508del-CFTR alleles are functionally non-equivalent. In other words, sequences adjacent to the F508del-CFTR mutation which are phylogenetically younger than the F508del variant and/or sequences in the vicinity of the CFTR gene allowing for allelic diversity among F508del chromosomes through recombination have a functional impact on the manifestation of the CF basic defect and disease severity. We would like to emphasise the point that variant F508del-CFTR genes (Praw=0.0009) more than ENaC variants (Praw=0.0695 at locus SCNN1B/SCNN1G; Praw =0.0183 at T663A in SCNN1A) determined the sensitivity of the CF nasal epithelium to amiloride in our study population (figure 3).
Host defence capabilities affect the manifestation of the CF basic defect
We selected cytokines as well as cytokine and pathogen receptors as candidate genes and tested these for an association with basic defect phenotypes. Minor (0.01<Praw<0.1; TLR5, CD14, IFNGR1, TLR4, TGFB1), moderate (0.001<Praw<0.01; IL1B, TLR9, TNFα, CD95, STAT3), and significant (0.0001<Praw<0.001; TNFR) associations of host defence genotype to basic defect phenotype were observed (figure 3). As none of these genes encode ion channels or their intermediate interaction partners, these genotype–phenotype relations were a priori unanticipated. However, crosstalk between the immune system and secretory properties of epithelial cells have been described before: for cytokines interleukin 10 (IL10), interferon γ (IFNγ), tumour necrosis factor α (TNFα), IL1β, transforming growth factor β (TGFβ), IL4 and IL13, alterations of CFTR and/or ENaC and/or CaCC expression and/or activity, resulting in altered ion and fluid transport, have been communicated.38–41 This has been interpreted as an unspecific host response to pathogens as the surface liquid is increased on the epithelium upon induction of a hypersecretory state by inflammatory stimuli,39 and consequently mucociliary clearance of the intruding pathogen will be alleviated. Our data show that the effect of the host defence system on ion conductance properties of CF epithelia outweighs the effect of major ion channels which we also analysed in our association study.
Discussion
Modifier genes have been studied in CF since Zielenski et al described CFM1 in 1999.23 Among those studies which analyse several candidate genes at once, one modifier only is reported.12 26 On the other hand, negative outcomes in these studies are often contradicted by other isolated reports (reviewed by Cutting24) and genetic modelling has led to the expectation that many more modifiers need to be uncovered in CF.4 25 This ambiguity is likely to be understood if non-inherited factors,1 6 which act differently between CF centres7 as well as between patients from different birth cohorts,5 are taken into account. Improvement of therapy, quality of life and life expectancy is at the heart of medical care for all diseases. However, the increase in lifespan among CF patients within the last decades is outstanding in comparison to other chronic diseases, signifying that better medication and improvement of therapeutic management dramatically alter the environment with which our patients have to cope. As a consequence, for the human geneticist studying a CF modifier in a cross-sectionally recruited patient sample, carriers of high risk alleles will be less frequent, especially among early born patient cohorts.20 Consequently, case–control comparisons might lack power to detect an allelic imbalance if the frequency of the risk alleles within the entire CF population is reduced owing to the survivor effect. If parental genotype information is available, the allelic bias introduced by a survivor effect can be visualised by comparing transmitted and non-transmitted alleles within CF families.18 In support of this, many loci reported in this article are positive in the transmission disequilibrium test (figure 3).
The impact of non-inherited factors on the course of CF disease, and moreover the change of these confounders over time, challenges classical strategies to validate findings of association studies. Ideally, replication studies with large sample sizes are used to verify findings. However, large CF patient samples are likely to be heterogeneous and thus will not share the major environmental determinants of CF disease progression. Taking this into account, we chose to replicate the findings of our association study using confirmatory CF endophenotypes and within-dataset validation strategies. No evidence for the role of the gene as a CF modifier was found for six loci. Isolated findings—defined as an association to only disease severity or only a basic defect phenotype—were observed for 20 genes. The remaining 26 loci analysed in this study are positive for both, an association with a clinical phenotype and a CF basic defect phenotype (see figure 3 and supplemental table 8). As the basic defect in CF is correlated with disease severity11 these consistent findings must be interpreted with caution with respect to the underlying causality. Either the gene analysed has a direct influence on the efficiency of F508del-CFTR processing and trafficking and thus causes a better phenotype; or alternatively, the patient subsamples analysed for CFTR mediated residual current in ICM or NPD can be considered as enriched with individuals who are mildly diseased due to the association of mild disease with residual function in respiratory and intestinal tissue.11 In the latter case, the findings of the association study do not necessarily imply a direct causal action of the analysed modifier on the CF basic defect, but can be interpreted as a supporting finding in two independent patient subsamples. As an example, the mild TNFR haplotype (allele 1-10-2-2 at markers rs767455, D12S889, rs1800692 and rs1800693)18 is significantly enriched on transmitted chromosomes in the entire European CF sibling population as indicated by TDT (Stanke et al,18 figure 3) and is associated with the concordant mildly affected phenotype (Stanke et al,18 figure 3) as well as with residual chloride secretion of the nasal epithelium (figure 4). As a causal interpretation, this might reflect the interaction of the cytokine pathway and ion secretory properties of the epithelium,38–41 promoting residual chloride secretion among carriers of the mild TNFR allele. Alternatively, as we observed residual chloride secretion more frequently among patients with mild disease phenotype,11 the TNFR association with the basic defect phenotype might reflect an overrepresentation of mild modifier alleles in this patient subgroup, which is equivalent to a replication study with confirmatory outcome.
In conclusion, describing modifiers in cystic fibrosis remains challenging even though the underlying disease is monogenic by definition. In contrast, the manifestation of endophenotypes such as diabetes, the CF basic defect and ultimately disease severity is most likely polygenic by nature, implying that when looking for CF modifiers, the same obstacles will be encountered as in complex diseases such as asthma or cardiovascular disorders. Based on our experience, we suggest that patient samples in which individuals share as many non-inherited characteristics as possible, and genetic markers which are selected for a high information content to overcome the loss of power in small sized study cohorts, will facilitate mapping of clinically meaningful CF modifiers in the future.
Acknowledgments
We are indebted to all participating patients and their families, physicians, CF centres and staff for their time, cooperation and assistance. The authors also thank Professor T Epplen (University of Münster) for his contribution of the genomic sequence for two microsatellites used to interrogate the HLA class I and III gene loci.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Funding VK was a member of the International Research Training Group Pseudomonas: Pathogenicity and Biotechnology (IRTG653 of the Deutsche Forschungsgemeinschaft). The support of the Fritz-Thyssen-Stiftung (to FS and TB) for analysis of the SCNN1B/SCNN1G-locus is gratefully acknowledged. MM Jr was supported by VZFNM00064203 and NE9488/3. This study was supported by a grant to BT from the Deutsche Forschungsgemeinschaft (SFB 621, project C7). This work was executed as part of the European Cystic Fibrosis Twin and Sibling Study and financially supported by the European Union (QL G1-CF- 2001-01005), the Deutsche Fördergesellschaft für die Mukoviszidoseforschung eV and the Mukoviszidose eV. The funding organisations had no influence on the study design, data analysis and interpretation or intention to publish.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was approved by the ethics committee of the Medizinische Hochschule Hannover.
Provenance and peer review Not commissioned; externally peer reviewed.