Article Text

Abstract
Background Hereditary short stature syndromes are clinically and genetically heterogeneous disorders and the cause have not been fully identified. Yakuts are a population isolated in Asia; they live in the far east of the Russian Federation and have a high prevalence of hereditary short stature syndrome including 3-M syndrome. A novel short stature syndrome in Yakuts is reported here, which is characterised by autosomal recessive inheritance, severe postnatal growth retardation, facial dysmorphism with senile face, small hands and feet, normal intelligence, Pelger-Huët anomaly of leucocytes, and optic atrophy with loss of visual acuity and colour vision. This new syndrome is designated as short stature with optic atrophy and Pelger-Huët anomaly (SOPH) syndrome.
Aims To identify a causative gene for SOPH syndrome.
Methods Genomewide homozygosity mapping was conducted in 33 patients in 30 families.
Results The disease locus was mapped to the 1.1 Mb region on chromosome 2p24.3, including the neuroblastoma amplified sequence (NBAS) gene. Subsequently, 33 of 34 patients were identified with SOPH syndrome and had a 5741G/A nucleotide substitution (resulting in the amino acid substitution R1914H) in the NBAS gene in the homozygous state. None of the 203 normal Yakuts individuals had this substitution in the homozygous state. Immunohistochemical analysis revealed that the NBAS protein is well expressed in retinal ganglion cells, epidermal skin cells, and leucocyte cytoplasm in controls as well as a patient with SOPH syndrome.
Conclusion These findings suggest that function of NBAS may associate with the pathogenesis of short stature syndrome as well as optic atrophy and Pelger-Huët anomaly.
- Short stature
- Yakuts
- Pelger-Huët anomaly
- optic atrophy
- NBAS
- diagnosis
- endocrinology
- clinical genetics
- haematology (incl blood transfusion)
- ophthalmology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Short stature
- Yakuts
- Pelger-Huët anomaly
- optic atrophy
- NBAS
- diagnosis
- endocrinology
- clinical genetics
- haematology (incl blood transfusion)
- ophthalmology
Introduction
Hereditary short stature syndromes are clinically and genetically heterogeneous disorders characterised by growth retardation with characteristic dysmorphism in the face and body. Although the various genes have been identified as being causative for hereditary short status syndrome, many cases are not explained. Yakuts are a population isolate in Asian and live in the northeastern part of Siberia in the Republic of Sakha of the Russian Federation.1 2 They exhibit high frequencies of some hereditary disorders.3 Among them short stature syndrome, a rare recessive inherited disease in other populations, is the major hereditary disease.4 There are two types of short stature syndrome in Yakuts. We applied the homozygosity mapping approach and subsequently identified an identical mutation in the Cullin 7 (CUL7) gene in all 43 Yakut patients with the short stature syndrome, indicating that Yakuts have a founder chromosome responsible for the CUL7 mutation.1 2 5 The CUL7 gene has been identified as a causative gene for 3-M syndrome (MIM 273750), which is a rare autosomal recessive disorder characterised by severe pre- and postnatal growth retardation and facial dysmorphism but with normal intelligence.2 6 7 The prevalence rate of short stature syndrome with CUL7 mutation in Yakuts is approximately 0.01% (at least 43 patients among 440 000 people), whereas only fewer than 60 patients with 3-M syndrome have been reported thus far.7–15 This result confirms that Yakuts are a population isolate. The other type of short stature syndrome in Yakuts is an autosomal recessive postnatal growth failure, normal intelligence, loss of visual acuity, and Pelger–Huët anomaly (PAH), which is characterised by an abnormal nuclear shape in neutrophil granulocytes (MIM 169400). In this study, we mapped the disease locus for this new type of short stature syndrome to 2p24.3 and identified an identical missense mutation in the neuroblastoma amplified sequence (NBAS) gene in patients with this syndrome.
Patients and methods
Patients and control samples
In the regional Department of Medical Genetical Consultation of the Republican Hospital No. 1-National Medical Centre in Yakutsk (Russia), we enrolled patients from 31 Yakut families who fulfilled the following clinical criteria: short stature, autosomal recessive inheritance, and optic nerve atrophy. To exclude the possibility that they had mutations in CUL7, which results in Yakut short status syndrome, we sequenced the coding region in CUL7 and found no nucleotide substitution. Ophthalmological examination was performed by paediatric ophthalmologists. Affected siblings were recruited from three families. We retrospectively reviewed the medical records of these patients. Birth size and subsequent lengths/heights were expressed as standard deviation scores (SDS) according to the Yakut standards. For analysis of PHA, we calculated the average lobe index (ALI) of the neutrophils using the following formula: the total number of nuclear lobes in 100 neutrophils divided by 100.16 We used 203 DNA samples from healthy Yakut individuals as normal controls. Genomic DNA was extracted from peripheral blood leucocytes according to a standard protocol. Blood samples were obtained after patients gave informed consent. The study was approved by the institutional review board of Niigata University.
Genome wide homozygosity mapping
We performed genome wide homozygosity mapping of the 33 affected individuals using 811 microsatellite markers (ABI PRISM Linkage Mapping Set HD-5; Applied Biosystems, Foster City, California, USA) covering the entire autosome with an average interval of 4.6 cM. We carried out polymerase chain reaction (PCR) using various MapPair microsatellite markers and analysed the PCR products using an ABI Prism 3100 genetic analyser (Applied Biosystems). We determined all allele sizes using the GeneScan (version 3.1.2; Applied Biosystems) and GENEMAPPER programs (Applied Biosystems). The genetic distance between adjacent markers was determined with the Marshfield sex averaged linkage map.
Fine homozygosity mapping in the candidate region
To narrow the candidate region, three dinucleotide polymorphic markers, M1491, M1542, and M1599 between D2S219 and D2S2295, were developed on the basis of simple repeat information from the UCSC Genome Browser on Human dated May 2004 (http://genome.ucsc.edu/index.html). The positions of chromosome 2 on each marker are 14913070 bp, 15429187 bp, and 15991354 bp, respectively. The following primer pairs were designed for these new markers: M1491 (forward primer, 5′- AGTTTTGCTAACCTGGAAGTC-3′ and reverse primer, 5′- ctgtgttccatcttctatgtg-3′); M1542 (forward primer, 5′- CATGTGCACTCACATGAATAC -3′ and reverse primer, 5′-CTGGCCAGTATACCTAATTTG-3′); and M1599 (forward primer, 5′- AATCCAAGGTCTGAGAGCAG-3′ and reverse primer, 5′-GCAGTGGACATCATCCAATC-3′). PCR reactions consisted of an initial denaturation step of 3 min at 95°C, amplification with denaturation for 30 cycles of 1 min at 95°C, annealing for 1 min at 55°C, and extension for 1 min at 72°C, followed by a final extension step for 10 min at 72°C. The allele frequencies of each marker were determined by analysing 50 Yakut control subjects.
Mutational analyses of DDX1 and NBAS genes
A series of 24 intronic primers for amplifying the 26 coding exons of DDX1 and a series of 52 intronic primers for 52 coding exons of NBAS were designed (see supplementary table). Each exon was amplified by PCR and the amplified products were purified with ExoSAP-IT (Amersham-Bioscience, Sunnyvale, California, USA) to a cycle sequence reaction with the use of BigDye Terminator version 3.0 (Applied Biosystems). We purified the reaction products using a NucleoSEQ kit (Macherey-Nagel, Düren, Germany) and analysed the products using an ABI 3100 DNA sequencer (Applied Biosystems). PolyPhen (http://genetics.bwh.harvard.edu/pph/) was used to predict the effect of the substitutions.17
Immunohistochemical analysis
Normal human tissue slides were obtained from US Biomax (Rockville, MD), Inc. Anti-NBAS mouse polyclonal antibody (Abnova Corporation, Taipei, Taiwan) was used for immunostaining. To assess the expression style of the antigen of the neuroblastoma amplified sequence (NBAS) in normal humans, we prepared formalin fixed, paraffin embedded sections of eye (82-year-old woman, 58-year-old man, 1-year-old girl, and 1-year-old boy), brain tissue (73-year-old woman), and skin (50-year-old woman). Peripheral blood smears were fixed at 20% formalin. Eye and brain tissue sections were pretreated with heat retrieval in a microwave oven for 30 min in 10 mM sodium citrate buffer (pH 6.0). These samples were immunostained by the avidin-biotin-peroxidase complex (ABC) method with a Vectastain ABC kit (Vector, Burlingame, California, USA) and mouse polyclonal antibody against NBAS (1:150; Abnova Corporation). Diaminobenzidine was used as the chromogen. For lamin B receptor (LBR) immunostainig, peripheral blood smears were fixed at 4% paraformaldehyde for 10 min, followed by immunostaining with rabbit monoclonal antibody against LBR (1:800; ab32535; Abcam, Cambridge, UK) and Alexa 568-conjugated goat anti-rabbit secondary antibody (1:1000; Molecular Probes, Eugene, Oregon, USA).
Results
Clinical features of a novel short stature syndrome
We enrolled 22 (64.7%) female and 12 (35.3%) male patients. The length of gestation varied from 38 to 40 weeks (median, 39 weeks); all patients were born by normal delivery. The SDSs of height by age from birth to 30 years is shown in figure 1. The average SDS of height at birth was −0.94±0.29 in females, 0.31±0.42 in males, and −0.65±0.26 in total (mean±SD). Although the SDS of height was within 1 SDS at birth, after 1 year of age, the average SDS of height was −4.44±1.19 in females, −3.16±1.06 in males, and −4.01±1.29 in total (mean±SD). The growth retardation was more severe in females than in males (p<0.001) (figure 1). There was no statistically significant correlation between age and SDS (r=−0.76, p=0.386) for older than 1 year.

Body length standard deviation scores (SDS) by age in 34 Yakut patients with short stature with optic atrophy and Pelger–Huët anomaly (SOPH) syndrome. Body length SDS were evaluated in 22 female (open circles) and 12 male (closed triangles) patients of different ages. Twenty-two female and 10 male patients of different ages were evaluated more than two times. Red bars represent mean body length SDS.
The clinical characteristics of 34 patients are presented in table 1. Ninety-seven per cent (33 cases) of the patients had normal intelligence. Eighty-eight per cent of the patients had a brachycephalic skull with hypoplasia of frontal and parietal tubers and a flat occipital area of the head (figure 2A, B and E). The patients had a characteristic face with straight nose, prominent glabella, small orbit, slight bilateral exophthalmos, hypoplastic cheekbone, narrow forehead, long philtrum, and thin lips (figure 2A, B, E and F). The patients' tissue turgor and skin elasticity were decreased in some patients (figure 2F, G and H). Size of the head was proportional for the body length (figure 2B, G and H), whereas length of the four limbs was relatively short for the body length. Their hands and feet were small (micromelia) (figure 2C and D). Their feet were wide and small, accompanied by a high arch and sandal chink (about 90%). Brachydactyly was observed in all patients (100%) (figure 2C and D). Male hypogonadism was not observed. The size of the uterus and ovaries of the female patients was normal, except in four individuals. One affected woman has a son of normal height.
Clinical features at first diagnosis in 34 Yakut patients with short stature with optic atrophy and Pelger–Huët anomaly (SOPH) syndrome
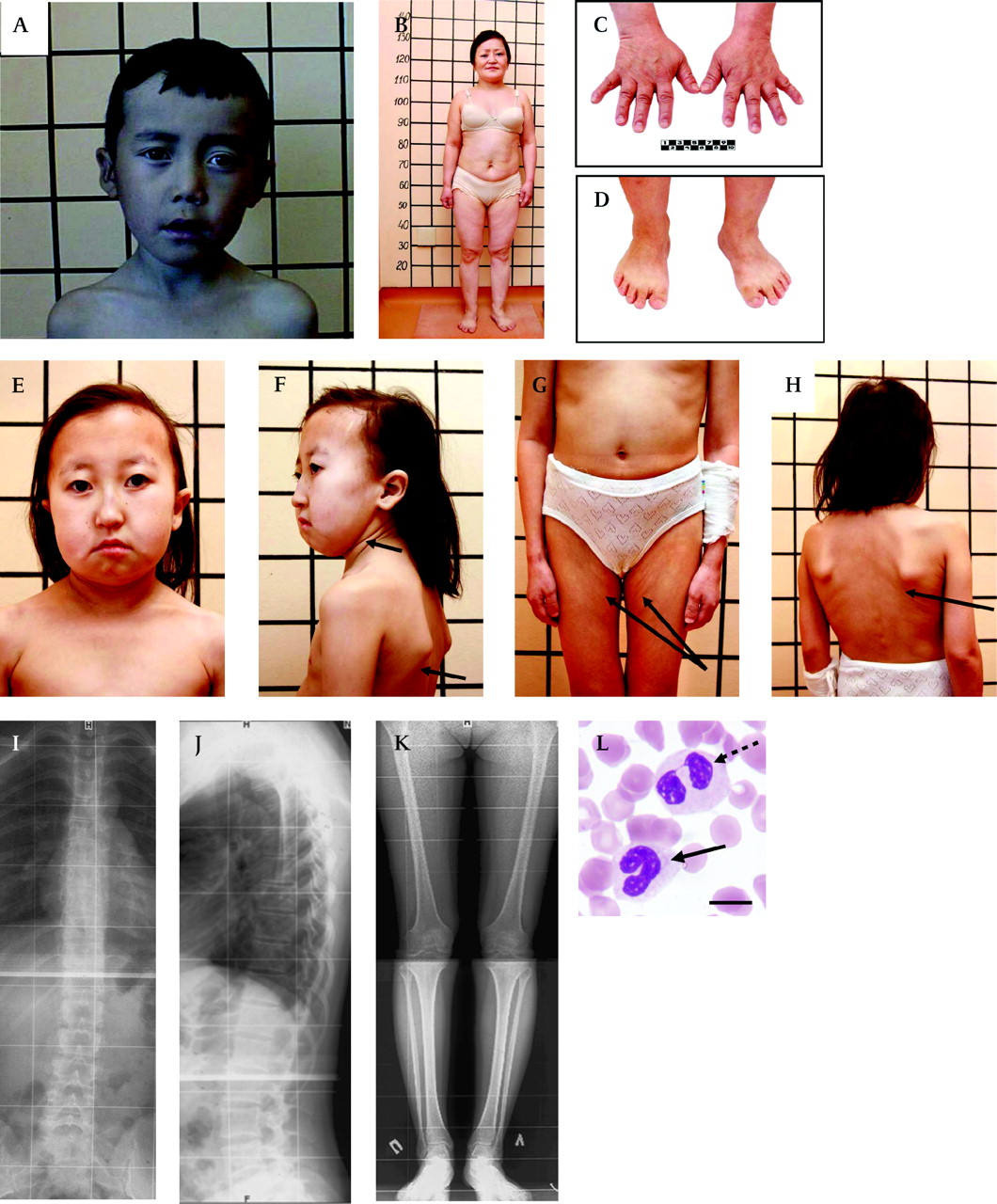
Clinical features of patients with short stature with optic atrophy and Pelger–Huët anomaly (SOPH) syndrome. (A) 10-year-old boy with SOPH syndrome. Note the brachycephalic skull, narrow forehead, long senile face with asymmetry, straight nose with prominent glabella, thick eyebrows, slight bilateral exophthalmos, left external strabismus, hypoplastic cheekbone, narrow forehead, long philtrum and thin lips. (B) Woman (33 years old) with SOPH syndrome. Note the short proportional stature, short neck, short and wide thorax, short upper and lower extremities. The lines behind patients denote body length (cm). (C, D) Brachydactyly and small hands (C) and feet (D) of patient B. (E) 12-year-old girl with SOPH syndrome. Note brachycephalic skull with fine hair, small features of face, senile face with asymmetry, thin lips, short neck. (F, G, H) The patients' tissue turgor and skin elasticity are decreased (arrows). (I, J) Radiograph of the thoracolumbar spine of a 21-year-old woman with SOPH syndrome shows osteochondrosis of thoracolumbar spine in lateral (I) and frontal (J) projection. (K) Radiograph of the lower extremities of a 21-year-old woman with SOPH syndrome shows osteoporosis. (L) Blood smear shows a homozygous neutrophil for SOPH syndrome with a non-segmental (arrows) and bilobed nucleus (dashed arrows). Scale bar indicates 10 μm.
Bone aging of the patient's hands was delayed in 14 patients (41.2%). Four cases in five patients examined by spinal x-ray showed abnormal findings, including: late ossification of vertebral apophysis (one case), dysplastic changes of spine and hypoplasia of the 12th ribs (one case), osteochondrosis thoracic and lumbar spine (two cases) (figure 2I and J), osteoporosis (two cases) (figure 2K), and dysplastic changes in low extremities (two cases).
Growth hormone provocation test with insulin was normal in all examined patients (13 cases). Serum free thyroxine was normal in all examined patients (28 cases). Brain magnetic resonance images (MRIs) were obtained for 24 patients and showed slight cerebellar atrophy (three cases), Dandy–Walker malformation (one case), cyst at internal capsule (one case), and empty selae (one case). All of the examined patients (22 cases in 21 families) had a high frequency of hypolobulation of granulocyte nuclei, indicating that they had PHA (figure 2M). The ALIs of the neutrophils were 3.75±0.01 in two normal Yakut individuals, 3.03±0.35 in four carriers, and 1.52±0.35 in 11 patients (mean±SD).
One of the patients' initial symptoms was visual loss (figures 3A, B, C, D, E and F). The onset of visual loss was 4.3±1.4 years of age (mean±SD; n=34). All patients had bilateral optic nerve atrophy (100%) and non-progressive impairment of visual acuity (0.23±0.21 (mean±SD; n=25)). Disc paleness with different degree of and expanded excavation was observed on the fundi (figure 3A). Borders of the optic disc were clear, and arteries and veins were normal. None of the patients had visual field defect, central scotoma, or nystagmus. All examined patients11 had great difficulty reading any of the Ishihara pseudoisochromatic plates (24 plate edition, 1964). We also performed the Farnsworth panel D-15 test on these 11 patients and found that one patient made a tritan error. We obtained electric sensitivity threshold and labiality from 30 patients. The values were 245.00±101.34 mkA for electric sensitivity threshold and 25.20±3.49 Hz for labiality, indicating an impairment of functions of the retina and the optic nerve. Myopia (12 cases), strabismus (nine cases), and hypermetropia (three cases) were also observed.
On the basis of these distinct clinical features, we named this new type syndrome as short stature with optic atrophy and Pelger–Huët anomaly (SOPH) syndrome.
Identification of the disease locus to chromosome 2p24.3
Because Yakuts are a population isolate, we applied the homozygosity mapping approach.1 5 A genome wide homozygosity mapping revealed that 25 of the 33 patients shared a four allele at D2S149 in the homozygous state: the frequency of the four allele in the normal Yakut population was 0.06 (figure 4A). Next we conducted fine genotyping using additional flanking microsatellite markers in this region (ie, D2S168, D2S328, D2S2199, D2S2267, D2S219, D2S312, D2S2295, D2S1360, and D2S2375). We found that all patients had an identical allele at D2S312 in the homozygous state (figure 4A). Loss of homozygosity was observed distal to marker D2S219 and proximal to marker D2S2295. Then we carried out further genotyping using microsatellite markers located between D2S219 and D2S2295 (ie, M1491, M1542, and M1599); 26 patients shared the identical haplotype 1-3-3-1 at M1491, D2S312, M1542, and M1599 in the homozygote state. With regard to patient 817, we could not exclude the possibility that this patient was a compound heterozygote. Loss of homozygosity was observed distal to marker M1491 and proximal to marker M1599 (figure 4A). Patient AA3362 showed loss of homozygosity at M1542. However, the patient had 1/1 genotype at M1599 that is identical to those of the other patients, suggesting that this could be accounted for by replication slippage with the marker M1542. Taking these findings together, we concluded that the causative gene for SOPH is located in a 1.1 Mb segment between M1491 and M1599 (figure 4B).

Ocular findings of 15-year-old boy with short stature with optic atrophy and Pelger–Huët anomaly (SOPH) syndrome. A 15-year-old boy (case 20) had poor vision and photophobia. His best corrected visual acuities were 20/40 in both eyes with mild myopic astigmatism. (A) Fundus photograph of the right eye. Optic disc showed mild simple optic atrophy. Although the macula and the peripheral retina were normal in appearance, the central retina was a little brownish with arteriole narrowing around the optic disc, suggesting nerve fibre layer defect of the retina. Arterioles were narrowed toward the optic disc (reverse tapering), some of which appeared to be associated with white sheathing (arrows). (B) Analysis by optical coherence tomography (OCT) (Optovue, RTVue-100, Fremont, California, USA) in the right eye. Upper figure depicts thickness of retinal nerve fibre layer (RNFL) along a 2 mm radius from the centre of the optic disc measured by OCT. (Nerve Head Map program: the NHM4 scan). Three significant levels, p>5%, p< 5%, and p<1%, are represented in green, yellow, and red as shown in the legend box. An RNFL defect was seen in significantly superior, temporal, and inferior directions, but the nasal direction was spared. Average thickness of the RNFL was significantly thin (69.31 μm, p<p0.01). Lower figure depicts thickness of the inner retinal layer. Significance of damage of the ganglion cell complex consisting of the RNFL, the ganglion cell layer, and the inner plexiform layer is shown in the colour map. Severe damage is seen around the fovea (grey circle). Average thickness of the ganglion cell complex is significantly thin (62.89 μm, p<p0.01). Fluorescent angiography of the retina revealed no abnormality. (C) The panel D-15 test (Luneau, Cedex, France). The patient revealed typical tritan colour defect. Colour vision was obviously affected. The patient could not read the Ishihara pseudoisochromatic plates. (D) Humphrey perimetry (Carl Zeiss Meditec, Dublin, California, USA). The central 30-2 threshold program test of the right eye showed slight general depression; mean deviation is –6.78 dB (p<0.5%) and pattern SD is 3.15 dB (normal). (E) Blue-on-yellow perimetry with the same instrument used in E. Blue spot stimulus (1.72°) with a peak transmission at 440 nm was used against a yellow background (wavelengths of 530 nm, luminance of 100 cd/m2). The central 30-2 threshold program test of the right eye showed significant and generalised depression; mean deviation was –19.32 dB (p<p0.5%) and pattern SD is 4.41 dB (normal). (F) Electroretinogram (ERG) findings. Findings of the right eye are aligned in the left column to compare with the normal pattern in the left column. Although a significant response from rod cells (scotopic dim flashes) was evident, the response from cone cells (photopic bright flashes and 30 Hz flicker ERG) was severely reduced, indicating that cone cells were severely affected selectively. Oscillatory potentials were decreased in both eyes. The ERG procedure complied with the International Society for Clinical Electrophysiology of Vision standard protocol.33 The recording procedures are described in a previous report.34
Identification of nucleotide substitution in NBAS gene in SOPH syndrome
In the critical region, there were two genes: neuroblastoma-amplified sequence gene (NBAS, MIM 608025) and DEAD (asp-Glu-Ala-Asp) box polypeptide 1 (DDX1) gene (figure 4B). We performed sequence analysis of NBAS and DDX1 in the patients using a series of intronic primers to amplify the coding exons and exon–intronic junctions in the NBAS and DDX1 genes. We found no mutation in DDX1, whereas we identified six nucleotide substitutions in the NBAS gene in the homozygous state in 33 of 34 patients; 130C→G (resulting in the amino acid substitution Q44E) in exon 2; 1611A→G (not altering the amino acid sequence) in exon 16; 2775T→C (not altering the amino acid sequence); 2845G→C (resulting in the amino acid substitution V949L) in exon 25; 3026G→C (resulting in the amino acid substitution C1009S) in exon 26; and 5741G→A (resulting in the amino acid substitution R1914H) in exon 45 (table 2). To exclude the possibility of the presence of heterozygous deletions, we analysed both parents in 10 families and found true homozygosity in all cases. Among these families, in family 17, the unaffected father of patient 414 had five of these six nucleotide substitutions, except 5741G→A (R1914H), in the homozygous state (figure 5A). Furthermore, arginine at position 1914 is highly conserved among species, whereas glutamine at position 44, valine at position 949, and cysteine at position 1009 were not conserved among species (figure 5B). Among these substitutes, only R1914H substitution was predicted to be possibly damaging by the Polyphen program. None of the 203 Yakut normal controls had the 5741G→A allele in the homozygous state. One hundred Japanese normal controls did not have the 5741G→A allele. Although patient 817 had these nucleotide substitutions in the heterozygous state, we concluded that homozygous 5741G→A substitution in the NBAS gene associates with the SOPH syndrome.
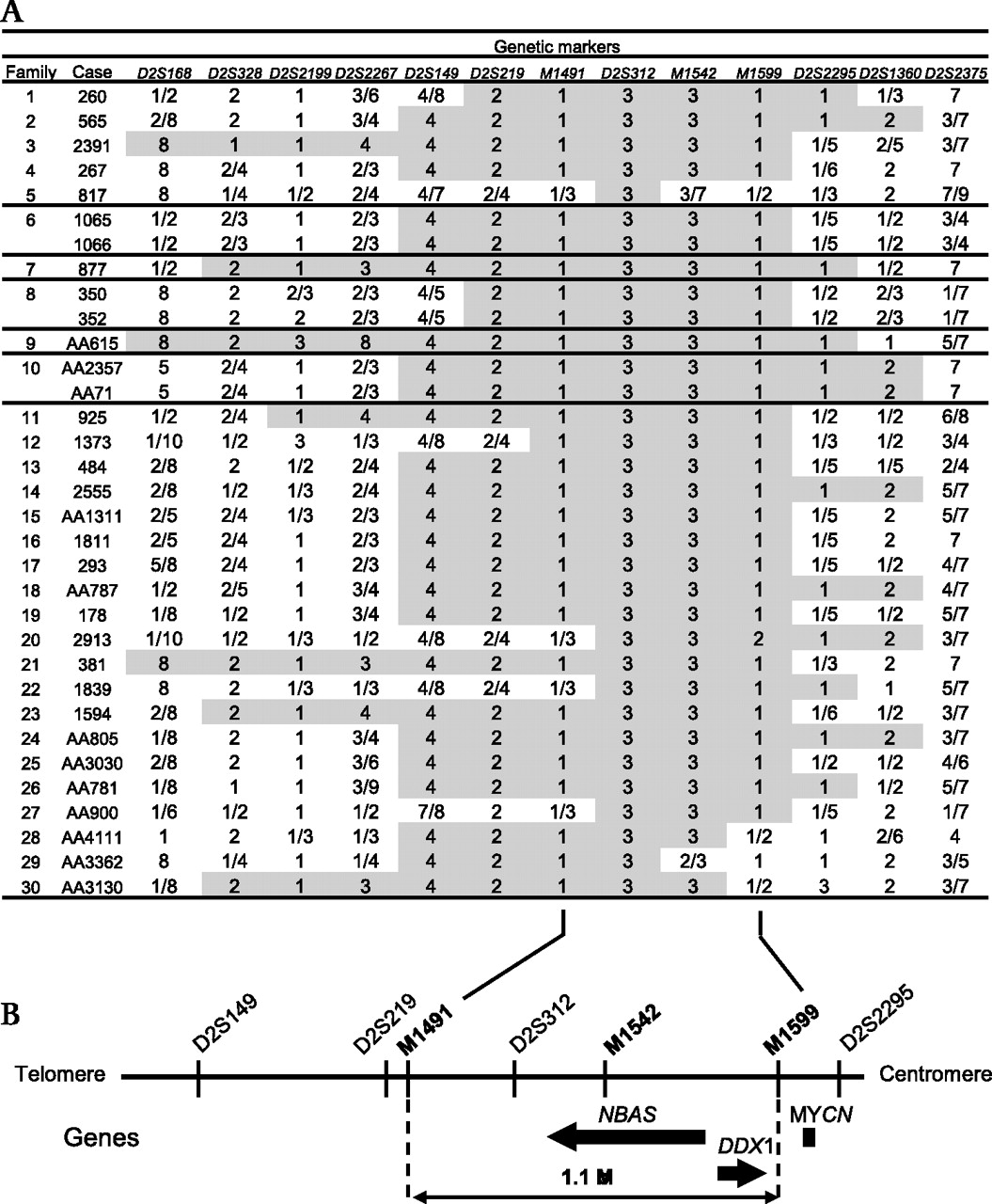
Genotype of 30 families with short stature with optic atrophy and Pelger–Huët anomaly (SOPH) syndrome for markers on 2p24.3. (A) All the patients have homozygous genotype (3/3) at D2S312. Homozygous genotypes around D2S312 are shaded in grey. (B) Physical map for 2p24.3. NBAS and DDX1 exist in the critical region defined by M1491 and M1599.
Polymorphisms in NBAS gene identified in patients with short stature with optic atrophy and Pelger–Huët anomaly (SOPH) syndrome
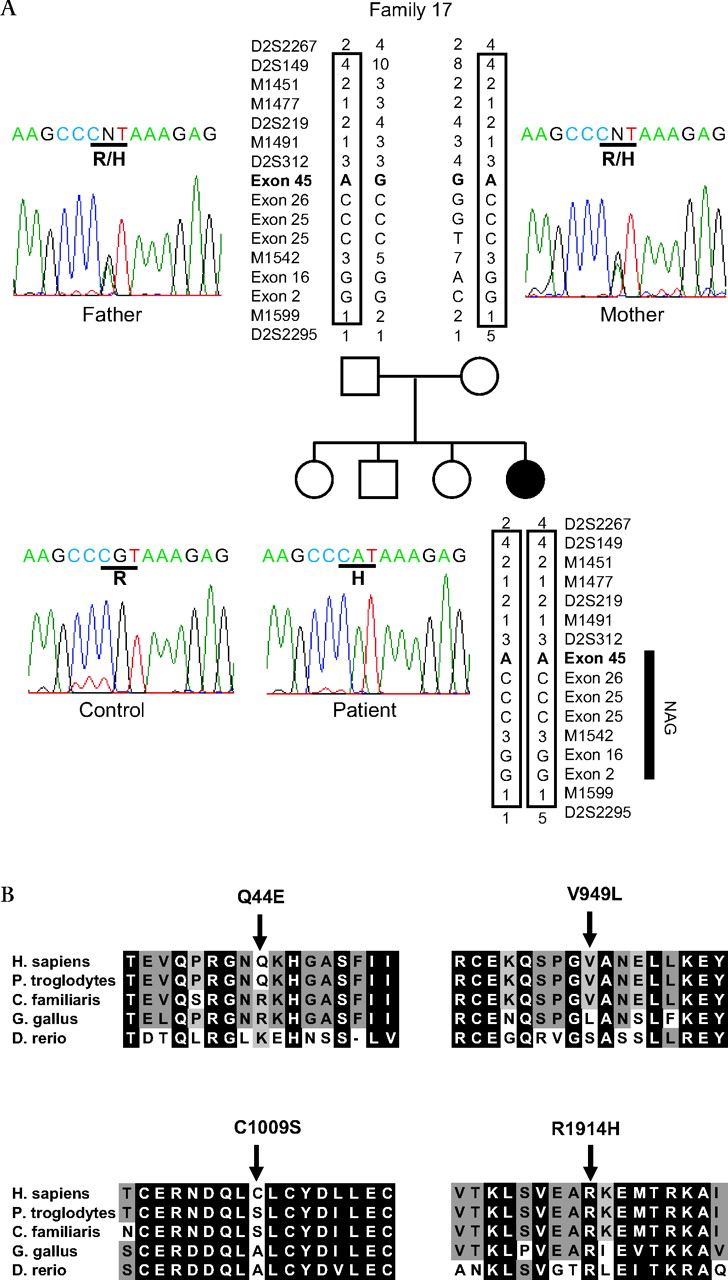
Haplotype of NBAS mutant chromosome and multiple alignment of NBAS (neuroblastoma amplified sequence) protein. (A) Haplotype of NBAS mutant chromosome for family 17 with short stature with optic atrophy and Pelger–Huët anomaly (SOPH) syndrome and chromatograms of the mutation in the NBAS gene. Disease associated haplotype is boxed. Five nucleotide substitutions, 130C→G (resulting in the amino acid substitution Q44E) in exon 2; 1611A→G (not altering the amino acid sequence) in exon 16; 2775T→C (not altering the amino acid sequence); 2845G→C (resulting in the amino acid substitution V949L) in exon 25; and 3026G→C (resulting in the amino acid substitution C1009S) in exon 26 in NBAS, were observed in a patient and her father in the homozygous state. 5741G→A (resulting in the amino acid substitution R1914H) in exon 45 alteration is observed only in the patient. (B) Amino acid sequence alignment of Homo sapiens NBAS ( GenBank accession number NP_056993. 2) with orthologs from Pan troglodytes (XP_001161679.1), Canis familiaris (XP_540088.2), Gallus gallus (XP_419959.2), and Danio rerio (NP_001038272.1) revealed by the ClustalW program (http://www.ebi.ac.uk/Tools/clustalw/). Conserved amino acid residues are shaded (GeneDoc program, http://www.nrbsc.org/gfx/genedoc/). Each shade represents a degree of conservation (black, 100%; dark grey, 80%; and grey, 60%). The mutated amino acid R1914H was highly conserved from human to fish, whereas Q44E, V949L, and C1009S were less conserved than R1914H. An amino acid homology search was conducted using the standard protein–protein BLAST. An amino acid sequence of human NBAS ( GenBank accession number NP_056993. 2), chimpanzee NBAS (XP_001161679.1), dog NBAS (XP_540088.2), chicken NBAS (XP_419959.2), and fish NBAS, (NP_001038272.1) were multiply aligned by the ClustalW program, version 1.83 with default parameters.
Immunohistochemical analysis for NBAS protein
In the retina, the cytoplasm of retinal ganglia cells and some of the inner layer cells was stained with NBAS antibody (figure 6A), whereas the outer layer cells and optic nerve were not stained with NBAS antibody (figure 6B). NBAS was also expressed in the cytoplasm of squamous epidermal cells (figure 6C); the expression was comparable between a patient and a control (figure 6C and 6D). With regard to leucocytes, immunostaining for NBAS showed comparable expression in leucocyte cytoplasm of normal individuals and patients with SOPH. The autosomal dominant inherited PHA is caused by heterozygous null mutations in the LBR gene.18 Because the amount of LBR quantitatively affects the lobulation of neutrophic nuclei, we investigated expression of the LBR in affected individuals.18 The expression of LBR protein, however, was comparable between normal individuals and patients (figure 6G and H).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immunohistochemical analysis of NBAS (neuroblastoma amplified sequence) protein. (A) Retina of autopsied control subject (58-year-old man) shows expression of an NBAS protein in the cytoplasm of ganglion cells. The scale bar indicates 20 μm. In the retina, the cytoplasm of retinal ganglia cells and some of the inner layer cells are stained with NBAS antibody (figure 6A), whereas the outer layer cells and an optic nerve are not stained with NBAS antibody (figure 6B). (B) NBAS is not expressed in cerebral optic nerve tract from autopsied control subject (75-year-old woman with myositis). The scale bar indicates 200 μm. (C, D) Cytoplasm of epidermal squamous cells are well stained with an NBAS antibody in a subject with short stature with optic atrophy and Pelger–Huët anomaly (SOPH) (C: 14-year-old boy) and a control (D: 50-year-old woman). The scale bar indicates 50 μm. (E, F) Cytoplasm of neutrophils are well stained with an NBAS antibody in a subject with SOPH (E: 14-year-old boy) and a control (F: 36-year-old man). The scale bar indicates 10 μm. (G, H) Neutrophils are well stained with a LABR antibody both in a subject with SOPH (G: 14-year-old boy) and a control (H: 36-year-old man). The scale bar indicates 5 μm.
Discussion
In this study, we have established a new disease that is a type of short stature syndrome with autosomal recessive inheritance. The presentation of loss of visual acuity and PHA (MIM 169400) is unique and renders the clinical phenotype distinguishable from other short stature syndromes, including 3-M syndrome. The cardinal features of this syndrome are postnatal growth retardation, small hands and feet, and loss of visual acuity and abnormalities of colour vision. Furthermore, blood smears of affected individuals showed PHA. In this study, we mapped the locus for this new type of short stature syndrome to 2p24.3 and identified an identical missense mutation in the NBAS gene. Thirty-three of 34 patients with SOPH syndrome have 5741G→A (R1914H) substitution in the NBAS gene in the homozygous state. The arginine at position 1914 is highly conserved among species, and Polyphen program suggested R1914H to be possibly damaging. None of the 203 Yakut had the 5741G→A allele in the homozygous state. None of the 100 Japanese normal controls had the 5741G→A allele. These observations indicate that 5741G→A (R1914H) allele is associated with the SOPH syndrome. One patient had a single 5741G→A substitution in the heterozygous state. The simplest explanation for these cases is that we failed to detect the second mutation, which may be located in regulatory domains of the NBAS gene that are yet to be defined. Further analysis is necessary to elucidate the disease mechanism for patient 817.
The prevalence rate of SOPH syndrome in Yakuts is 1 per 10 000 (at least 33 patients among 440 000 people); the carrier frequency of the 5741G→A substitution of NBAS is approximately 1% in the Yakut population. Furthermore, all of the affected individuals have other rare nucleotide substitutions in NBAS: 130C→G in exon 2; 1611A→G in exon 16; 2775T→C and 2845G→C in exon 25; and 3026G→C in exon 26. The results indicate the Yakuts have a founder chromosome responsible for the SOPH syndrome. We have previously demonstrated that all Yakut patients with 3-M syndrome have an identical mutation in CUL7.2 Taken together, these results strongly indicate that Yakuts are a population isolate. Because Yakuts are the first population isolate discovered in Asia, this population would play an important role in identifying a susceptibility gene for complex diseases in people with an Asian background.19
The NBAS gene was identified as a gene that is co-amplified with the N-myc (MYCN) gene in neuroblastoma cell lines.20 Northern blot analysis has revealed two alternative transcripts: a 4.5 kb transcript encoding a deduced 1353 amino acid protein and a 7.3 kb transcript encoding a deduced 2371 amino acid protein.20 21 The longer transcript, with a predicted molecular weight of 268 kDa, is generally more abundant. NBAS messenger RNA is highly expressed in connective tissues, eye, brain, and spinal cord, suggesting that NBAS protein may play an essential role in these organs (expression profile is available from NCBI: http://www.ncbi.nlm.nih.gov/UniGene/, S.O.U.R.C.E: http://source.stanford.edu/cgi-bin/source/sourceSearch). Although the function of NBAS remains unknown, it has two putative leucine zippers, a ribosomal S14 motif and a domain homologous to secretary pathway protein in yeast, Sec39.22 23 Sec39 is required for vesicle transport from the endoplasmic reticulum (ER) to the Golgi apparatus and from the Golgi apparatus to the ER. Indeed, recently, NBAS has been identified as a member of a subunit of the syntaxin 18 complex, which is associated with membrane transport; depletion of NBAS causes redistribution of Golgi recycling proteins accompanied by a defect in protein glycosylation, indicating that NBAS is involved in Golgi-to-ER transport.24 It remains unclear why mutation of NBAS results in the narrowly restricted clinical phenotypes in those with SOPH. NBAS might play an important role for Golgi-to-ER or vesicle transport in the retinal ganglion cells and affected organs. In a patient with SOPH syndrome, the expression of NBAS was well preserved in skin and leucocyte. Therefore the dysfunction of NBAS resulting from the R1914H substitution may underlie the pathogenesis of the SOPH syndrome.
Non-progressive loss of visual acuity accompanied by optic nerve atrophy is a characteristic clinical feature in SOPH syndrome. NBAS was expressed in ganglion cells in the adult retina. Although we did not obtain retina from the patients with SOPH, these findings suggest that SOPH comprises inherited optic neuropathies that resulted from loss of retinal ganglion cells.25 Leber's hereditary optic neuropathy (MIM 535000) and optic atrophy 1 (MIM 165500) are the most frequent hereditary optic neuropathies. The function of the causative genes for these optic neuropathies is tightly associated with mitochondrial function. Therefore, it would be interesting to investigate the function of NBAS on mitochondria in ganglion cells.
Analysis of the electroretinogram (ERG) for the individual with SOPH syndrome revealed that the cone cell pathway was also involved in SOPH syndrome. The visual and colour impairment for patients with SOPH syndrome is a relatively stationary disorder described as ‘cone dysfunction’, not progressive cone dystrophy.26 Most of the patients with SOPH syndrome had complete achromatopsia (total colour blindness); however, one of these individuals showed the blue-yellow deficiency (tritan). These results indicate that the homozygous state of 5741G→A substitution in the NBAS gene does not lead only to complete achromatopsia but can also cause incomplete achromatopsia. Phenotype variability in cone dysfunction syndrome has also been reported in patients with CNGA3 mutation.27
Another characteristic finding in SOPH is PHA, which is characterised by a hyposegmented nuclear shape and a coarse chromatin organisation in neutrophils. PHA is characterised by an abnormal nuclear shape in neutrophil granulocytes. The mutation in the LBR gene, which is located at 1q42.1, has been identified as a causative gene for PHA.18 Although this disorder is autosomal dominant, Pelger–Huët-like cells have also been observed in many pathological conditions (eg, myelodysplasia and myeloproliferative disease), certain drugs, and occasional acute infections.28 An autosomal dominant PHA is caused by heterozygous null mutations in the LBR gene.18 LBR protein is embedded within the inner nuclear membrane that interacts with B-type lamins and nucleosomes. Although the expression of LBR is reduced in cells with PHA,29 we were unable to find a difference in LBR expression in leucocytes from patients with SOPH syndrome. The NBAS gene is the second gene that results in PHA and the first gene that results in PHA in an autosomal recessive manner. Although there are no data to suggest that NBAS protein associates with nuclear matrix proteins, a physiological role for NBAS will clarify the molecular mechanism of PHA.
Interestingly, PHA has occasionally been described in association with short status syndrome.18 30 Hydrops–ectopic calcification–‘moth eaten’ (HEM)/Greenberg skeletal dysplasia, which is an autosomal recessive chondrodystrophy with PHA, is caused by homozygous mutations in the LBR.31 Moreover, rabbits with PHA homozygosity had microphthalmia and severe chondrodystrophy with limb defects.32 Although the skeletal deformity is not severe in SOPH syndrome, the association between the nuclear morphology in neutrophils and the development of the skeletal system is interesting to indicate the molecular link between nuclear membrane and skeletal development.
In conclusion, we have shown comprehensive clinical features of a new type of short stature syndrome prevalent in Yakuts and identified the missense mutation in the NBAS gene. This information may be useful for genetic counselling in the Yakut population. These results provide new insights for understanding the molecular mechanism for short stature syndromes, optic nerve atrophy, and PHA.
Acknowledgments
We sincerely thank the patients and family members for their participation, and M Tsuchiya and M Moriyama for technical assistance.
References
Supplementary materials
Web Only Data jmg.2009.074815
Files in this Data Supplement:
Web Only Data jmg.2009.074815
Files in this Data Supplement:
Footnotes
NM and KH contributed equally to this work.
Funding This study was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas ‘Advanced Brain Science Project’ and ‘Applied Genomics’, a grant for Young Researchers within the framework of the Program of the Japan-Russian Youth Exchange and HUGO Grant travel awards, and a Grant for the Promotion of Niigata University Research Projects.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the institutional review board of Niigata University.
Provenance and peer review Not commissioned; externally peer reviewed.