Article Text

Abstract
Background: Nephronophthisis (NPHP), a rare recessive cystic kidney disease, is the most frequent genetic cause of chronic renal failure in children and young adults. Mutations in nine genes (NPHP1-9) have been identified. NPHP can be associated with retinal degeneration (Senior-Løken syndrome), brainstem and cerebellar anomalies (Joubert syndrome), or liver fibrosis.
Methods: To identify a causative gene for the subset of patients with associated liver fibrosis, the authors performed a genome wide linkage search in a consanguineous family with three affected patients using 50K SNP microarrays and homozygosity mapping.
Results: The authors obtained a significant maximum parametric LOD (logarithm of odds) score of Zmax = 3.72 on chromosome 8q22 and identified a homozygous missense mutation in the gene MKS3/TMEM67. When examining a worldwide cohort of 62 independent patients with NPHP and associated liver fibrosis we identified altogether four novel mutations (p.W290L, p.C615R, p.G821S, and p.G821R) in five of them. Mutations of MKS3/TMEM67, found recently in Meckel–Gruber syndrome (MKS) type 3 and Joubert syndrome (JBTS) type 6, are predominantly truncating mutations. In contrast, the mutations detected here in patients with NPHP and associated liver fibrosis are exclusively missense mutations. This suggests that they may represent hypomorphic alleles, leading to a milder phenotype compared with the more severe MKS or JBTS phenotype. Additionally, mutation analysis for MKS3/TMEM67 in 120 patients with JBTS yielded seven different (four novel) mutations in five patients, four of whom also presented with congenital liver fibrosis.
Conclusions: Hypomorphic MKS3/TMEM67 mutations cause NPHP with liver fibrosis (NPHP11). This is the first report of MKS3 mutations in patients with no vermian agenesis and without neurological signs. Thus NPHP, JBTS, and MKS represent allelic disorders.
Statistics from Altmetric.com
Nephronophthisis (NPHP), an autosomal recessive cystic kidney disease, is the most frequent genetic cause of end stage renal disease. By positional cloning, nine causative NPHP genes have been identified so far.1 2 3 4 5 6 7 8 9 10 11 The characteristic histological findings in NPHP are renal interstitial infiltration with fibrosis, tubular atrophy with basement membrane disruption, and cyst development at the corticomedullary border. Identification of NPHP genes has contributed to a unifying pathogenic theory that describes cystic kidney diseases as “ciliopathies”. This implies that functional defects of primary cilia, basal bodies, and centrosomes are central to the pathogenesis of NPHP.12 The ubiquitous cilial expression of NPHP proteins might explain why other organs can also be affected in patients with NPHP. The most frequently associated extra-renal involvements are retinopathy described as Senior–Løken syndrome (15% of the cases), cerebellar ataxia known as Joubert syndrome (10–15%), and liver fibrosis (5%), also known as Boichis disease.13 14
JBTS is a developmental disorder characterised by brainstem malformation, cerebellar vermis hypoplasia/dysplasia, ataxia, hypotonia, mental retardation, neonatal breathing abnormalities, and oculomotor apraxia. Patients with additional clinical features including nephronophthisis, renal cystic dysplasia, hepatic fibrosis, ocular coloboma, retinal dystrophy, and polydactyly are referred to as cerebro-oculo-renal syndrome. To date, mutations in seven genes (AHI1, NPHP1, CEP290/NPHP6, MKS3/TMEM67, RPGRIP1L/NPHP8, ARL13B, and CC2D2A) have been identified in patients with JBTS.9 11 15 16 17 18 19 20 Similar multiple organ involvement was described in Meckel–Gruber syndrome (MKS), a perinatally lethal ciliopathy, which is characterised by central nervous system malformation (mainly occipital encephalocele), postaxial hexadactyly, hepatic bile duct proliferation with fibrosis, and multicystic dysplastic kidneys. It has been shown that the phenotypic spectrum of NPHP, JBTS, and MKS can be caused by mutations in the same gene. This applies to NPHP6/CEP290, and NPHP8/RPGRIP1L,11 18 as well as to NPHP1 which was shown to be allelic for NPHP and JBTS,16 and NPHP3 for NPHP and a MKS-like phenotype, respectively.21 Recently, allelism between JBTS and MKS has also been reported for ARL13B and CC2D2A.19 20 NPHP with associated liver fibrosis has been described as a distinct disease entity14; we wanted to identify the underlying causative gene by positional cloning.
Patients and methods
Patient data
Approval for human subjects research was obtained from the University of Michigan Institutional Review Board and the French ethical committee. We obtained blood samples, pedigree, and clinical information after receiving informed consent (www.renalgenes.org) from all patients and/or their parents. The diagnosis of NPHP was based on the following criteria: (1) clinical course with characteristic clinical signs of NPHP including chronic renal failure, polyuria, polydipsia, anaemia, and growth retardation; (2) renal ultrasound or renal biopsy compatible with the diagnosis of NPHP as judged by a (paediatric) nephrologist; (3) pedigree compatible with autosomal recessive inheritance. Neurological criteria for Joubert syndrome were based on the following clinical hallmarks of this cerebello-oculo-renal syndrome: (1) molar tooth sign (MTS) or (2) diagnosis of JBTS by a (paediatric) neurologist or geneticist. Associated JBTS symptoms were recorded: optic nerve or retinal coloboma, tapetoretinal degeneration. We excluded the presence of homozygous NPHP1 deletions in all patients by applying a multiplex polymerase chain reaction (PCR) approach.22
Homozygosity mapping
We used standard methods to isolate genomic DNA from peripheral blood samples according to the manufacturer’s instructions (Puregene, Gentra Systems, Minneapolis, Minnesota, USA).
From each patient, a DNA sample was processed according to the manufacturer’s instructions (Affymetrix GeneChip Human Mapping 100K Assay Manual). In brief, 250 ng of high quality genomic DNA was digested with HindIII and ligated to HindIII-adaptors. After PCR amplification, random fragmentation and labelling, samples were hybridised to the 50K array (GeneChip Human Mapping 50K Hind array; Affymetrix) in a hybridisation oven (Affymetrix Hybridisation Oven 640; Affymetrix, Santa Clara, California, USA). Washes and staining of the arrays were performed using a fluidics station (Affymetrix Fluidics Station 450; Affymetrix), and images were obtained using a gene-chip scanner (Affymetrix GeneChip Scanner 3000 Affymetrix). Call rates had to exceed 98%. Allele calls were made using the BRLMM (Bayesian Robust Linear Model with Mahalanobis distance classifier) algorithm. Data were evaluated by calculating multipoint LOD (logarithm of odds) scores across the whole genome using GENEHUNTER,23 assuming recessive inheritance with complete penetrance.
Mutation analysis
Mutation screening of MKS3/TMEM67 was performed by direct sequencing of all 28 exons and the adjacent intronic junctions using DNA samples of affected individuals of family F563 and F585. Primer sequences and PCR conditions are available upon request. PCR products were purified using spin columns according to the manufacturer’s instructions (Marligen, Ijamsville, Maryland, USA) and directly sequenced using the dideoxy chain termination method on an automatic capillary genetic analyser (Applied Biosystems, Foster City, California, USA). Segregation of the identified mutations was investigated in both families. Mutation screening in the remaining 60 patients with NPHP and liver fibrosis was performed using either heteroduplex based CEL I endonuclease single mismatch analysis (45 patients) or direct sequencing (15 patients). Preparation of the CEL I endonuclease, heteroduplex formation, and CEL I treatment were performed as described previously.22 Samples showing aberrant banding patterns were purified and directly sequenced. Additionally, we screened 120 patients with JBTS and cystic kidney involvement for mutations in MKS3/TMEM67 applying the CEL I endonuclease analysis. Furthermore, we examined 105 patients with NPHP without liver involvement. Out of these patients, we excluded 15 families showing no linkage to the MKS3 locus and analysed the remaining by direct sequencing (60) or by using the CEL I endonuclease prescreening method (30). Possible damaging effect of missense mutations was assessed by PolyPhen software.24
Haplotype analysis
Markers used for haplotype analysis consisted of microsatellite markers D8S88, D8S270, D8S1988, D8S1794, D8S1735, D8S1822, D8S1772, and D8S1129. Haplotype analysis was performed as described earlier.22
Results
Total genome search for linkage in patients with NPHP and liver fibrosis
The association of NPHP and liver fibrosis had been first described in 1973 as a distinct disease entity by Boichis et al and we see this association in about 5% of patients with NPHP. For three of these kindred with known consanguinity we performed a genome wide homozygosity mapping using 50K single nucleotide polymorphism (SNP) microarrays. Two of these kindred (F563, F585) showed an overlap of non-parametric LOD score peaks (NPL) on chromosome 8q that indicated homozygosity by descent (fig 1). In a consanguineous Turkish kindred with three affected individuals (F563), the parents were fourth degree cousins. In this kindred we detected two small regions of potential homozygosity by descent on chromosome 8q22.1. We obtained a significant maximum parametric LOD score of Zmax = 3.72 at marker SNP_A-1686154 (θ = 0) (fig 1A). In the other family, F585, 12 homozygous regions have been identified for the affected child, indicating homozygosity by descent (fig 1B). One of them overlapped on chromosome 8 with a homozygous region of family F563. The critical genetic interval of overlap spanned 1.03 Mb between heterozygous flanking markers SNP_A-1739376 (94,154,715 bp, University of California Santa Cruz (UCSC), Genome Browser; build hg18, March 2006) and SNP_A-1715464 (95,184,445 bp). From the six genes located within this interval we selected MKS3/TMEM67 as an excellent candidate gene, since mutations in this gene have been recently identified in patients with MKS (MKS3) and Joubert syndrome (JBTS6).18 25
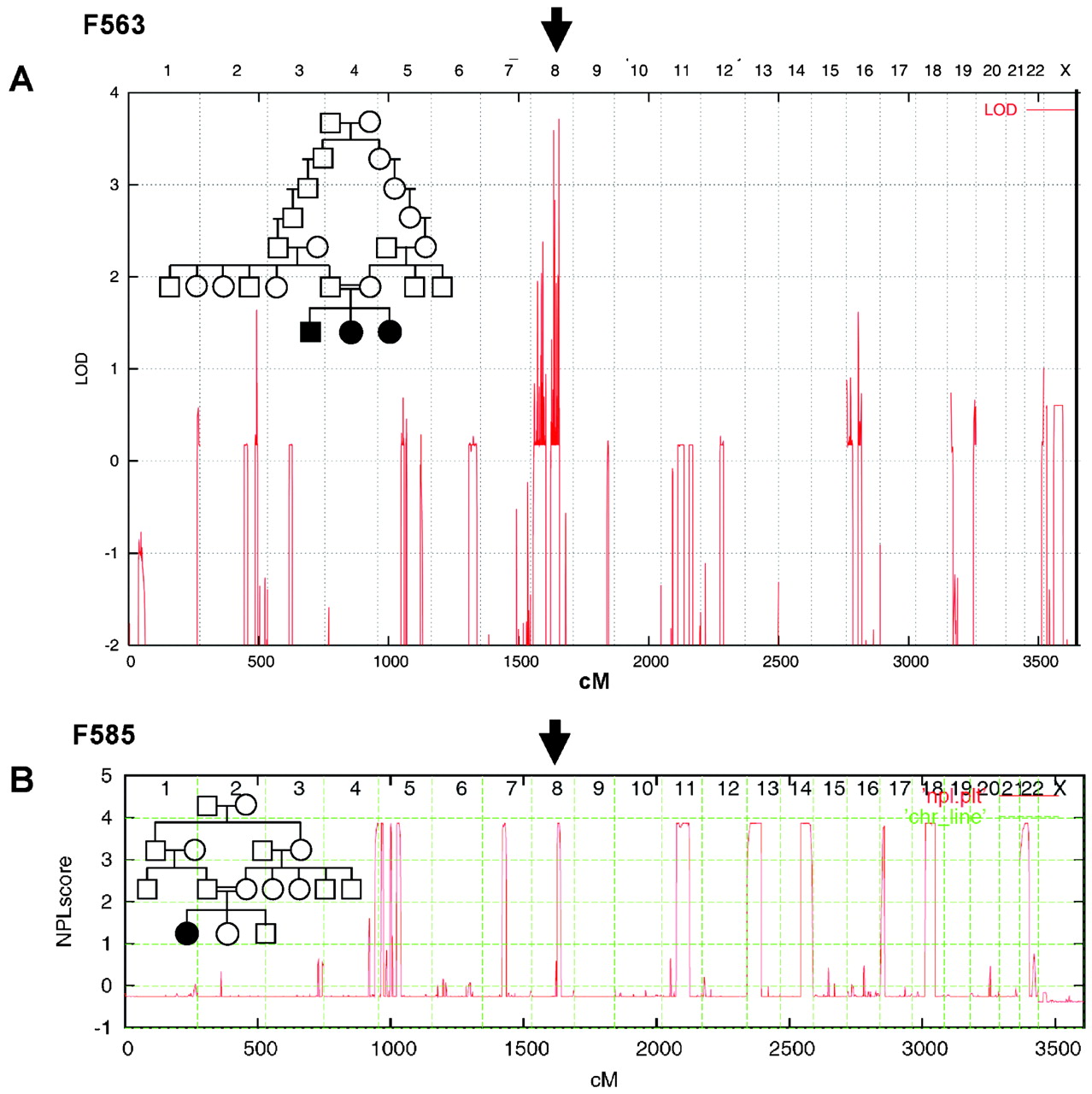
Total genome search for linkage by homozygosity mapping in two consanguineous Turkish kindred with nephronophthisis (NPHP) and liver fibrosis. (A) Graph represents parametric LOD (logarithm of odds) score on the y axis in relation to genetic position on the x axis for family F563. Human chromosomes are concatenated from 1p-ter (left) to 22q-ter (right) on the x axis. Genetic distance is given in cM. Note, that for family F563 with three affected siblings (parents are fourth degree cousins) a significant parametric LOD score of 3.72 was found on chromosome 8 (arrow) defining a new locus for NPHP (NPHP11). (B) Graph represents non-parametric LOD score for family F585. Parents of the affected child of kindred F585 are first degree cousins. NPL peaks (12) represent regions of putative homozygosity by descent, indicating candidate loci. The presence of an overlapping peak on chromosome 8q for F585 (arrow) with the peak of F563 further supports the presence of a putative locus for nephronophthisis with liver fibrosis.
Mutations in MKS3/TMEM67 in patients with NPHP and associated liver fibrosis
We carried out mutation analysis by direct sequencing of all 28 exons of MKS3/TMEM67 in families F563 and F585 and identified two different novel homozygous missense mutations (p.G821S, p.C615R) which segregated with the affected status (table 1, fig 2A,B).
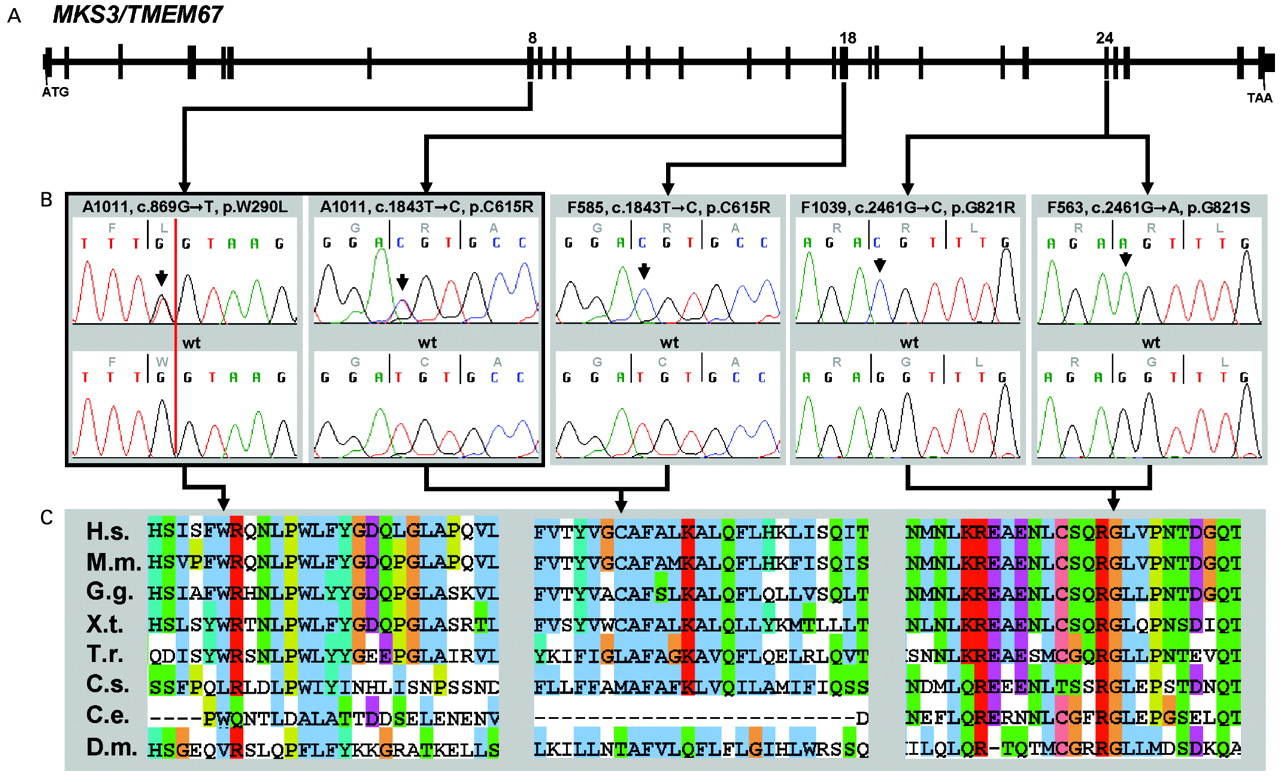
MKS3/TMEM67 mutations in patients with nephronophthisis (NPHP) and liver fibrosis. (A) Exon–intron structure of the MKS3/TMEM67 gene. Black boxes indicate 28 coding exons. (B) Sequencing chromatograms of four different MKS3/TMEM67 missense mutations detected in five families with nephronophthisis and associated liver fibrosis. Family number, amino acid sequence change, and mutated nucleotide are given above sequence traces. Wild-type (wt) sequences are shown below mutated sequences. (C) Alignment of the MKS3/TMEM67 amino acid sequence with homologs from various species. The amino acid sequences are aligned with those of Homo sapiens (H.s.), Mus musculus (M.m.), Gallus gallus (G.g.), Xenopus tropicalis (X.t.), Takifugu rubripes (T.r.), Ciona savignyi (C.s.), Caenorhabditis elegans (C.e), and Drosophila melanogaster (D.m.). Amino acid residues that are within the same chemical group are coded in the same colour. Positions of mutated amino acids are indicated with arrows.
Four different MKS3/TMEM67 mutations in five families with nephronophthisis (NPHP) and associated liver fibrosis
Both mutations were absent from at least 188 healthy control individuals and from an additional 147 ethnically matched healthy control samples from Turkey. Mutation screening in 60 additional patients with NPHP and associated liver fibrosis revealed two additional novel missense mutations (p.W290L, p.G821R) in three additional families who were homozygous or compound heterozygous for missense mutations (table 1, fig 2 A,B). The mutations p.W290L and p.G821R were absent from at least 91 healthy control individuals. The mutation p.G615R was identified in a patient of German origin (F519) and in a patient of Turkish origin (F585). Haplotype analysis using highly polymorphic microsatellite markers revealed a shared haplotype of six consecutive markers, indicating inheritance of the p.G615R mutation from a common ancestor (fig 3). Haplotype analysis in patients from German families F1039 and F56 who carry the TMEM67 mutation (c.2461G>C, p.G821R) revealed another shared haplotype indicating identity by descent (fig 3).

Pedigrees, segregation of MKS3/TMEM67 mutations, and haplotype sharing. The MKS3/TMEM67 mutation (c.1843T>C, p.C615R.) segregates in F585 with the affected status (the haplotypes segregating with the mutated allele is indicated as a black bar). Circles represent females; squares represent males; filled symbols denote patients with nephronophthisis or Joubert syndrome (F56). Haplotype analysis using eight highly polymorphic microsatellite markers (MS) on chromosome 8 surrounding the MKS3/TMEM67 locus revealed six shared consecutive markers among affected individuals of F585 and F519. This is consistent with potential identity by descent. Haplotype analysis in two patients from two different German families (F1039 and F56) with the MKS3/TMEM67 mutation (c.2461 G>C, p.G821R) revealed a shared haplotype of six consecutive MS markers (grey bars) indicating potential identity by descent. Note that F56 carries the mutation heterozygously and that the phase is inferred (parental DNA was not available).
Taken together, all three amino acids (W290, C615 and G821) mutated in patients with NPHP and hepatic fibrosis show evolutionary conservation to various degrees with G821 conserved throughout including Drosophila melanogaster and Caenorhabditis elegans (fig 2C), and their mutations were predicted by PolyPhen software program to be damaging with high position specific independ counts (PSIC) scores (p.W290L: 3.53, p.C615R: 3.22, p.G821S: 1.72, p.G821R: 2.17).
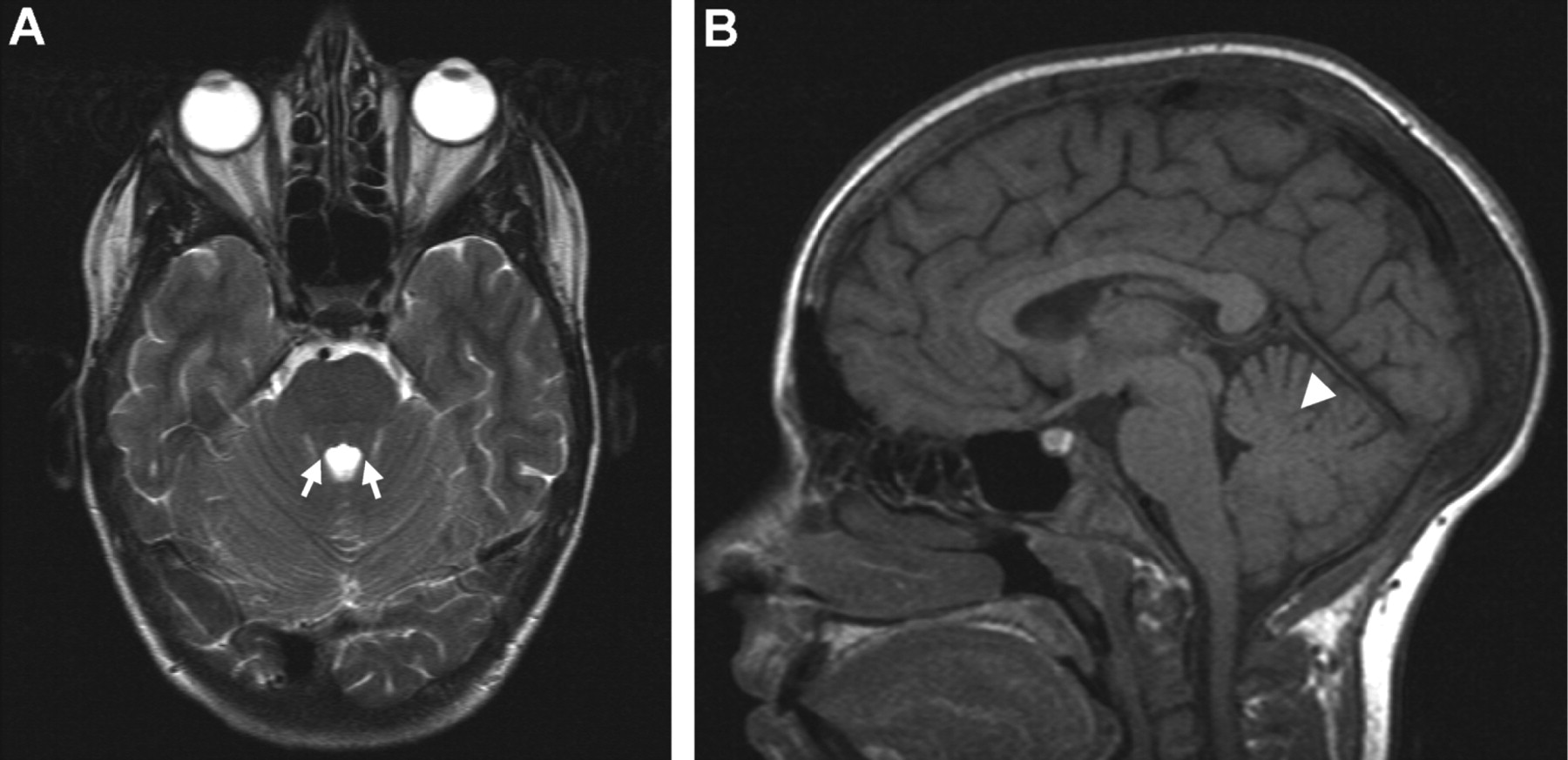
Patients had no or only mild neurological involvement and molar tooth sign was not found in any case with available brain magnetic resonance imaging (MRI) (table 1, fig 4). All patients developed end stage renal disease (ESRD) between the age of 6 and 14 years. Three patients had ocular symptoms.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Brain magnetic resonance images of a patient with nephronophthisis (NPHP) and liver fibrosis who carries an MKS3/TMEM67 mutation. Axial T2 (A) and midline sagittal T1 (B) images in patient F563-2. Note that the superior cerebellar peduncles (small white arrows) and the cerebellar vermis (white arrowhead) show no abnormalities.
Lack of MKS3/TMEM67 mutations in patients with NPHP without liver fibrosis
In order to investigate whether MKS3/TMEM67 mutations may also occur in isolated NPHP without hepatic involvement, we analysed 105 NPHP patients who presented without liver disease. After excluding 15 families not linked to the MKS3/TMEM67 locus we performed mutational analysis by direct sequencing of 60 samples and the remaining 30 samples by heteroduplex based CEL I endonuclease screening.22 No MKS3/TMEM67 mutation has been identified in this cohort.
Mutations in MKS3/TMEM67 in patients with Joubert syndrome and kidney involvement (NPHP)
We performed mutation screening in a total of 120 unrelated individuals with JBTS ascertained worldwide. Altogether, we identified recessive mutations in MKS3/TMEM67 in five patients with JBTS from four independent families (table 2).
Seven different MKS3/TMEM67 mutations in four families with Joubert/COACH syndrome
We found two nonsense mutations (p.Q44X, p.R208X) and five missense mutations (p.M252T, p.C615R, p.S630P, p.G821R, p.I833T). The mutations p.Q44X, pC615R, p.S630P, and p.G821R were novel findings. The missense mutations p.S630P and p.I833T were not present in >140 healthy control individuals and were predicted to be damaging by PolyPhen (PSIC score: 1.563 and 1.793, respectively). All four unrelated patients carried a missense mutation on at least one allele (table 2).
Consistent with the Joubert phenotype, all five patients presented with ataxia, hypotonia or psychomotor retardation or showed cerebellar vermis hypo- or aplasia (table 2). All patients developed ESRD between 8–15 years of age, consistent with juvenile nephronophthisis. Four of them were diagnosed with hepatic fibrosis. Importantly, similar to patients with MKS3/TMEM67 mutations without neurological involvement, four of them had ocular involvement, one sibling pair was blind, and retinal coloboma was found in three unrelated patients.
Discussion
The aim of this study was to identify a new gene implicated in NPHP and hepatic fibrosis. By positional cloning, we identified mutations in MKS3/TMEM67, a gene known to be implicated in Meckel–Gruber syndrome and Joubert syndrome. Screening a cohort of 62 independent patients with NPHP and associated hepatic fibrosis we found MKS3/TMEM67 mutations in five (8%) patients. Furthermore, we detected MKS3/TMEM67 mutations in four of 120 (3.3%) unrelated patients with JBTS and kidney involvement (nephronophthisis). Mutations in the putative transmembrane receptor meckelin (MKS3/TMEM67) were initially identified in fetuses with Meckel–Gruber syndrome.25 Recently, Baala and colleagues identified mutations in MKS3/TMEM67 in patients with Joubert syndrome (JBTS6) and Brancati and colleagues reported MKS3/TMEM67 mutations in patients with COACH syndrome.18 26 COACH is a distinct subgroup of Joubert syndrome defined by Cerebellar vermis hypo/aplasia, Oligophrenia, congenital Ataxia, Coloboma and congenital Hepatic fibrosis. Allelism between MKS, JBTS, and in some NPHP cases has been described for the genes CEP290/NPHP6, RPGRIP1L/NPHP8, ARL13B, and CC2D2A.9 11 19 20 27 Very recently, mutations in NPHP3 have been reported in fetuses with an MKS-like phenotype21 and allelism has been described. Hypomorphic mutations in NPHP3 are shown to lead to NPHP with or without hepatic involvement in children and adolescents. MKS3/TMEM67 mutations identified so far in MKS were in 71% (27/38 alleles) truncating mutations—that is, either frameshift, nonsense or obligatory splice site mutations as reviewed in supplemental table 1. Furthermore, 11 out of these 19 published families with MKS (58%) carry truncating mutations on both alleles, and only two fetuses carry solely missense mutations (supplemental table 1). One of these mutations (p.Q376P) was shown to have functional significance as the mutated meckelin protein was unable to localise at the cell membrane.28
To date, MKS3/TMEM67 mutations have been reported in 12 independent families with JBTS/COACH syndrome (supplemental table 1 available online).18 26 Taking also the four families with JBTS and MKS3/TMEM67 mutations presented within the present study into account, we find that none of the patients with JBTS6 carries truncating mutations on both alleles, with the exception of one family harbouring a homozygous in-frame deletion (Baala et al18). It should be noted that while truncating mutations on both alleles are associated with exencephaly and MKS, all patients with Joubert syndrome had at least one missense mutation. However, it does seem significant that all patients with NPHP and hepatic fibrosis and no brain anomaly carry a missense mutation affecting either amino acid C615 or G821. This provides novel insights into the genotype–phenotype correlation for two reasons. First, it shows that some hypomorphic mutations of MKS3/TMEM67 do not lead to any neurological impairment. Brain imaging, when available, indeed showed normal morphology of the brainstem and the cerebellum in these patients. Second, we present the first NPHP cases carrying MKS3/TMEM67 mutations, as fetuses with MKS3 and reported patients with JBTS6 developed cystic kidney dysplasia, rather than cystic nephronophthisis.18 25 29 30 Interestingly, we also found ocular involvement in seven out of 12 patients with MKS3 mutations with or without neurological involvement. These symptoms included retinal coloboma, retinal degeneration or oculomotor disorders. Ocular coloboma was reported in only one MKS fetus,29 and oculomotor apraxia in one patient with JBTS.18 However, these ocular abnormalities are probably underdiagnosed, especially in fetuses with MKS.
On the other hand, liver seems to be affected in most of the patients with MKS3 mutations independently of the neurological involvement. Enlarged liver, hepatic fibrosis and/or bile duct proliferation was reported in all published MKS3 cases with available information and in three out of five patients with JBTS6.18 We found liver fibrosis in all seven patients with NPHP but no neurological involvement and in all four JBTS6 patients with available information. Along the same line, we have not found MKS3/TMEM67 mutations in 105 NPHP patients without liver involvement. Liver disease thus seems to be a specific feature resulting from MKS3/TMEM67 mutations. Consequently, its mutation analysis should be considered in patients with NPHP and liver fibrosis. Nevertheless, its mutation rate is low, as we found MKS3/TMEM67 mutations in only five of 62 (8%) patients with NPHP and associated liver fibrosis indicating further heterogeneity.
We conclude that mutations in MKS3/TMEM67 can cause NPHP (NPHP11) in patients with additional liver fibrosis but without neurological involvement and normal brain imaging. Thus, NPHP (NPHP11), MKS (MKS3) and JBTS (JBTS6) represent a spectrum of allelic disorders.
Acknowledgments
We sincerely thank the patients and their families for participation. We are grateful to the following physicians for contribution of materials and clinical data from patients. Drube, Hannover, Germany; K Koenig, Berlin, Germany; Gaedicke, Berlin, Germany; Stolpe, Rostock, Germany; D Haffner, Rostock, Germany; L Stapenhorst, Cologne, Germany; O Mehls, Heidelberg, Germany, M Duval-Arnould, Creil, France. We thank Tania Attié-Bitach for providing the primers and Céline Becker for technical assistance.
REFERENCES
Supplementary materials
Web only appendix for 46;10:663
Files in this Data Supplement:
Footnotes
An additional table is published online only at http://jmg.bmj.com/content/vol46/issue10
EAO and KT contributed equally to this work
Funding This research was supported by grants from the National Institutes of Health to FH (DKRO1-069274, DKRO1-068306, DKRO1-064614). FH is the Frederick GL Huetwell Professor, a Doris Duke Distinguished Clinical Scientist, and an Investigator of the Howard Hughes Medical Institute. KT is supported by an ERA-EDTA fellowship.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.