Article Text

Abstract
Silver–Russell syndrome (SRS MIM180860) is a disorder characterised by intrauterine and/or postnatal growth restriction and typical facies. However, the clinical picture is extremely diverse due to numerous diagnostic features reflecting a heterogeneous genetic disorder. The mode of inheritance is variable with sporadic cases also being described. Maternal uniparental disomy (mUPD) of chromosome 7 accounts for 10% of SRS cases and many candidate imprinted genes on 7 have been investigated. Chromosome 11 has moved to the forefront as the key chromosome in the aetiology, with reports of methylation defects in the H19 imprinted domain associated with the phenotype in 35–65% of SRS patients. Methylation aberrations have been described in a number of other imprinted growth related disorders such as Beckwith–Wiedmann syndrome. This review discusses these recent developments as well as the previous work on chromosome 7. Other candidate genes/chromosomal regions previously investigated are tabled.
Statistics from Altmetric.com
The first clinical descriptions of Silver–Russell syndrome (SRS) were published by Silver et al1 and Russell2 who independently described unrelated children characterised by low birth weight and with additional features including triangular shaped face, pointed chin and body asymmetry in a subset of patients. Since then more than 400 cases have been reported in the literature with a variable estimated incidence from 1 in 3000 to 1 in 100 000.3 Throughout life, the manifestation of profound growth restriction persists and may lead to feeding difficulties with associated fasting hypoglycaemia. In general, this tends to be the main clinical problem and life expectancy appears to be unaffected by the condition. The main parental and clinical concern during infancy, therefore, is poor eating habits and much effort is expended in encouraging adequate feeding. For those children not showing catch up growth by the age of 2 years, growth hormone therapy is encouraged and is usually given daily as subcutaneous injections for up to 3 years.4 Price et al5 proposed that to be able to make a diagnosis of SRS, the patient must exhibit the following five key features as well as any number of the other features described in box 15 6:
Birth weight below or equal to –2 SD from population mean
Poor postnatal growth below or equal to −2 SD from the mean value at diagnosis
Preservation of occipitofrontal head circumference (OFC)
Classic facial features
Skeletal asymmetry.
GENETIC AETIOLOGY
SRS is a clinically and genetically heterogeneous disorder. Autosomal dominant, autosomal recessive and X-linked inheritance models have all been reported.7 Both concordant8 and discordant9 10 monozygotic twins for SRS have also been found. Given this complex genetic aetiology, it is not surprising that a number of different chromosomes and potential candidate genes have been implicated in the cause of SRS. Up to 2005, the most widely discussed chromosome involvement was with chromosome 7, but recent findings have shown that loci on chromosome 11 have a big part to play in the origins of this disorder. The implications of these exciting new findings are discussed in detail below.
Chromosome 7
Chromosome 7 as a candidate locus for SRS has been studied in detail after maternal uniparental disomy (mUPD) 7 was found in a subset of patients with pre- and postnatal growth restriction.11–14 It was postulated that the growth restriction might be due to growth related imprinted genes located on this chromosome.
Imprinted genes represent a very small proportion of the total gene complement, which differ from the rest in the manner of their transcriptional regulation. Unlike most genes that are active from both maternally and paternally inherited chromosomes (that is, biallelic), imprinted genes are only active from one parental allele (that is, monoallelic). For germ line imprinted genes, this is dictated in the egg or sperm. Imprinted genes are therefore regulated according to parent-of-origin, and function in a hemizygous state. An imprinted gene may be paternally expressed and maternally imprinted (silenced) or maternally expressed and paternally imprinted (silenced).
This hypothesis was elegantly demonstrated by pioneering work in the 1980s by Barton et al15 and McGrath and Solter16 who created diploid mouse embryos with either gynogenetic (completely maternal) or androgenetic (completely paternal) genomes to examine the phenotypic effect on conceptuses. Gynogenetic conceptuses produced only embryonic material with little or no placentation whereas androgenetic conceptuses displayed poor embryonic development with recognisable trophoblast tissue and yolk sacs. It was obvious from these experiments that the maternal and paternal genomes made differential contributions to fetal growth and development. This hypothesis was further defined by the creation of mouse strains with Robertsonian and reciprocal translocations to create artificial UPDs for sub-chromosomal regions. Interestingly, some chromosomes in the UPD state had no visible phenotypes whereas others showed profound growth related effects.17 18 One of the many cited examples is that of mice with maternal or paternal UPD for part of mouse chromosome 11, which is syntenic with human chromosome 7. Mice with mUPD11 were on average 30% smaller than their wild type littermates, independent of sex, while the pUPD11 mice were 30% larger than their wild type littermates.17 This demonstrated a polarising epigenetic effect on the same phenotype. It is now accepted that, in general, paternally expressed genes show promotion of fetal growth whereas maternally expressed genes have a tendency to restrict fetal growth. This imprint, which effectively silences one parental copy of a gene, is epigenetic, and is reset during gametogenesis.19
Box 1: Characteristics of Silver–Russell syndrome (SRS)
Physical features
Facial dysmorphism: classic facial features of small triangular facies, high forehead with pointed chin and down turned corners of the mouth
Short stature persisting into adulthood
Skeletal asymmetry
Preservation of occipitofrontal circumference
Clinodactyly
Camptodactyly ± distal arthrogryposis
Syndactyly of second and third toes
Hemihypertrophy
Urogenital dysmorphism:
Hypospadias
Posterior urethral valves
Inguinal hernia
Clinical features
Birth weight ⩽ −2 SD from the mean. Usually followed by failure to thrive
Poor head control during infancy due to relatively large head compared to body as well as general motor dysfunction due to lack of muscle bulk and strength
Feeding difficulties in infancy and childhood leading to fasting hypoglycaemia
Increased sweating during infancy
Gastrointestinal symptoms including gastro-oesophageal reflux, oesophagitis and food aversion
Developmental delay which may be associated with mental compromise in a subset of patients
Delayed bone age
Cardiac defects
Malignancies
Information adapted from OMIM entry 180860-http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id = 180860;5 6
The findings that maternal UPD7 in humans is associated with pre- and postnatal growth restriction, and that imprinted genes have a phenotypic dosage effect in mice, suggested chromosome 7 as a possible candidate for SRS pathogenesis. Several groups therefore screened cohorts of clinically defined SRS patients both for mUPD7 and/or imprinted gene(s) on chromosome 7 that may contribute to the observed growth disorder.20–22 UPD7 is readily detectable using polymorphic short tandem repeat (STR) microsatellite markers by polymerase chain reaction (PCR) or variable number of tandem repeat (VNTRs) by Southern blotting. Of the 98 SRS patients collectively screened by these groups, nine were found to have mUPD7 and it is now widely acknowledged that this mechanism is responsible for approximately 10% of SRS cases. As an alternative to disrupting imprinted gene expression, the hypothesis that the UPD might be uncovering a recessive allele was also tested.22 Several examples of this have been described for chromosome 7 where patients with cystic fibrosis appear to be homozygous for delta508 despite only having one carrier parent.11 12 23 This turns out to be due to isodisomy, where the patient inherits two copies of the same chromosome from one parent while the normal chromosome is excluded. Preece et al22 therefore analysed five mUPD7 patients for common regional isodisomic inheritance using 40 polymorphic markers distributed along the length of chromosome 7 with an average distance between markers of <10 cM. They found no common region of isodisomy among the five probands, suggesting the phenotypic effect of chromosome 7 is either due to exposure of recessive alleles for several different genes or much more likely due to a common imprinting effect. The observation of mUPD7 in SRS as a genetic aetiology appears to be an almost exclusive phenomenon as it has not yet been reported in screening of individuals with intrauterine growth restriction (IUGR) or pre-and/or postnatal growth restriction.24
Interestingly, Hoglund et al25 and Pan et al26 describe single case reports each of complete paternal isodisomy for chromosome 7 with normal growth phenotype. This contrasts sharply with the overgrowth phenotype seen in the syntenic pUPD11 mice and suggests that the human phenotype is related to maternal growth restricting genes rather than paternal growth promoting genes. However, a single case report of a boy with cystic fibrosis with complete paternal isodisomy for chromosome 7 displaying overgrowth was recently published, reopening the possibility of paternally active imprinted genes on this chromosome playing a role in growth.27 Two key candidate regions on chromosome 7 have been delineated as the most likely areas to contain the genes that may be causative factors in SRS.
7q32
Kobayashi et al28 suggested the human equivalent of the imprinted mouse Peg1/Mest (mesoderm expressed transcript) gene as a candidate for SRS. They developed a novel subtraction–hybridisation method to isolate imprinted genes systematically. Of eight paternally expressed genes (Pegs), Peg1/Mest was identified as the most abundantly expressed in embryos, specifically in the mesoderm.29 Mest maps to mouse proximal chromosome 6, which shares conserved synteny with human chromosome 7q21-qter. This region contains the paternally expressed MEST gene, which was the first imprinted gene to be identified on chromosome 7.28 Based on its imprinting status and the mouse phenotype, MEST has been investigated extensively as a candidate for SRS, but sequencing and methylation studies have as yet found no evidence for an involvement.30
An SRS patient with a rare segmental mUPD for the region 7q31-qter further implicated this region to be harbouring a potential candidate gene(s) for SRS.31 32 Recently, a case report of a girl born after assisted reproductive technology (ART) and diagnosed with SRS was investigated for methylation status of the MEST region.33 They observed partial hypermethylation of the MEST DMR and speculated that this may have some effect on this paternally expressed growth promoting gene.
Several other imprinted genes were found in the vicinity of MEST, including the paternally expressed intronic transcript from the biallelically expressed gamma-2 COP (nonclathrincoatprotein) (COPG21T1), paternally expressed MEST1T1 (also known as PEG1-AS) and the maternally expressed carboxypeptidase A4 (CPA4).34 CPA4 has a role in cell proliferation and differentiation.35 However, no mutations were seen for CPA4 in the 10 SRS patients screened or in other imprinted genes in this region.36 37 Neighbouring genes flanking these imprinted genes were all shown to be biallelic, delineating the extent of a small imprinting cluster.38 Meanwhile, mice either targeted for mutation of the homologous Mest gene or mice with mUPD for chromosome 6 (which are equivalent to null for the paternally expressed Mest gene), result not only in IUGR and reduction in postnatal survival but also in abnormal maternal behaviour including impaired placentophagia.39 40 The latter finding provided the first evidence that normal and abnormal imprinting may also play an important role in behaviour.
7p11.2-p13
Joyce et al41 reported a mother and daughter with some but not all of the key features of SRS. Cytogenetic and fluorescent in situ hybridisation (FISH) established that both mother and daughter had an inverted duplication of the proximal short arm of chromosome 7 encompassing the region 7p13-p12.1. This region harbours the growth factor receptor bound protein-10 (GRB10) gene which is imprinted in an isoform and tissue specific manner. GRB10 encodes a cytoplasmic adaptor protein which may facilitate linkage of cell surface tyrosine kinase receptors, such as the insulin receptor (IR) and insulin-like growth factor-1 receptor (IGFIR) to an uncharacterised mitogenic signalling pathway, and also binds epidermal growth factor receptor (EGFR) and the platelet derived growth factor receptor.
The orthologous Grb10 gene is located on mouse chromosome 11 which is associated with the growth imprinting phenotype described by Cattanach and Kirk.17 The GRB10 gene could be considered a very strong candidate for SRS.42 Monk et al43 identified a maternally derived de novo duplication in a female patient with some of the classic features of SRS. The duplication involved 7p13-p11.2, encompassing GRB10 at 7p12.2 and the biallelically expressed insulin-like growth factor binding proteins 1 and 3 (IGFBPI and IGFBP3).44 In addition to this family, a further two maternally inherited duplications encompassing GRB10, and several chromosomal disruptions in the 7p11.2-p13 region, have also been described.45 More recently a maternally inherited duplication of 7p12-p13 in a patient with cognitive deficit without features of SRS has been reported in the literature.46 This duplication does not extend to include GRB10 and delineates an SRS critical interval of 2.2 Mb containing only GRB10 and the biallelically expressed cordon bleu gene (COBL). Therefore the growth restriction observed is most likely due to the over-expression of the imprinted GRB10 gene.
The GRB10 gene is imprinted in a highly isoform and tissue specific manner. In both mouse and human, expression in the brain is derived from the paternal allele. In the mouse all other isoforms in embryonic and placental tissues are expressed from the maternal allele. This maternal expression is not completely conserved in the human, with only the γ1 and γ2 isoforms imprinted in muscle, and expression in other all tissues reported, derived from both parental alleles.47–49 Mice carrying maternally inherited targeted deletions of Grb10 show a growth phenotype identical to the phenotypes described for the pUPD11 and PatDp11 mice, consistent with an involvement in SRS.50 In total, 76 SRS patients were screened for mutations in GRB10 but no pathogenic changes were found.48 51 Arnaud et al49 and Monk et al52 found no significant changes in the differentially methylated region (DMR) for GRB10 in 24 and 22 SRS patients, respectively. Therefore, despite the location and proposed function of GRB10 and its function in mice, there have not been any conclusive reports of SRS being caused by over-expression or loss of imprinting of GRB10, or by gain of function coding mutations.
Chromosome 11
The 11p15.5 region contains two clusters of imprinted genes each regulated by its own imprinting centre region (ICR). H19 and insulin-like growth factor 2 (IGF2) are controlled by the H19 differentially methylated domain (DMD), which is the telomeric ICR1, while the genes within the KCNQ1 cluster are regulated by the centromeric KvDMR1 called ICR2. This region has become a key focus in the aetiology of SRS ever since the report of hypomethylation of the ICR1 in five of nine SRS patients studied by Gicquel et al.53 They hypothesised that epigenetic dysregulation of genes in the human 11p15.5 region encompassing the telomeric ICR1 region and the more centromeric ICR2 region involved in the aetiology of Beckwith–Wiedemann syndrome (BWS) (OMIM 130650) could be genotypically and phenotypically opposite for SRS.
BWS is characterised by macroglossia, pre- and/or postnatal growth greater than the 90th centile and abdominal wall defects.54 In addition, umbilical abnormalities and characteristic linear indentation of the earlobe are included in the diagnostic criteria. BWS patients are also at increased risk of tumours.55 56 Interestingly, these patients have large placentae that are almost twice the normal weight.57
The genetic aetiology of BWS is extremely heterogeneous with most reported cases being sporadic. In a study of 106 sporadic BWS cases, 17% were due to paternal isodisomy of the 11p15 region which contains the reciprocally imprinted genes IGF2 and H19 (ICR1) as well as the KCNQ1 domain (ICR2).58 As IGF2 is a potent prenatal growth factor, over-expression of IGF2 should lead to tissue hyperplasia.
Weksberg et al59 examined skin fibroblasts in five monozygotic twin pairs discordant for BWS and found loss of methylation of the maternal allele of the ICR2 in the affected twin only. Imprinting defects of the ICR2 that result in loss of expression of the cyclin dependent kinase inhibitor 1C (CDKN1C) gene are the most common epimutation found in 50% of BWS patients. Maternally inherited coding mutations of CDKN1C have also been reported in 5% of BWS patients.60 However, 10% of BWS patients exhibit hypermethylation of ICR1 and the promoter region, resulting in loss of expression of the non-coding H19 gene and the biallelic expression of IGF2.58
Research into the imprinting control of this domain reveals a complicated regulatory mechanism. The ICR1 is a paternally methylated germline DMR, located 2–4 kb upstream of the H19 transcript that contains several CCCTC binding factor (CTCF) binding sites. The binding of CTCF confers ICR1 function as a methylation sensitive insulator between the multiple IGF2 promoters and enhancers located downstream of H19. On the unmethylated maternal allele, CTCF binds to form a boundary that prevents the IGF2 promoters interacting with the enhancers, whereas on the methylated paternal allele, CTCF cannot bind and the IGF2 promoters can freely associate with the enhancer to bring about expression from the paternal allele only.61 62 Therefore any gain or loss of methylation of ICR1 will result in aberrant IGF2 expression, potentially corresponding to the opposite growth phenotypes of SRS and BWS (fig 1).
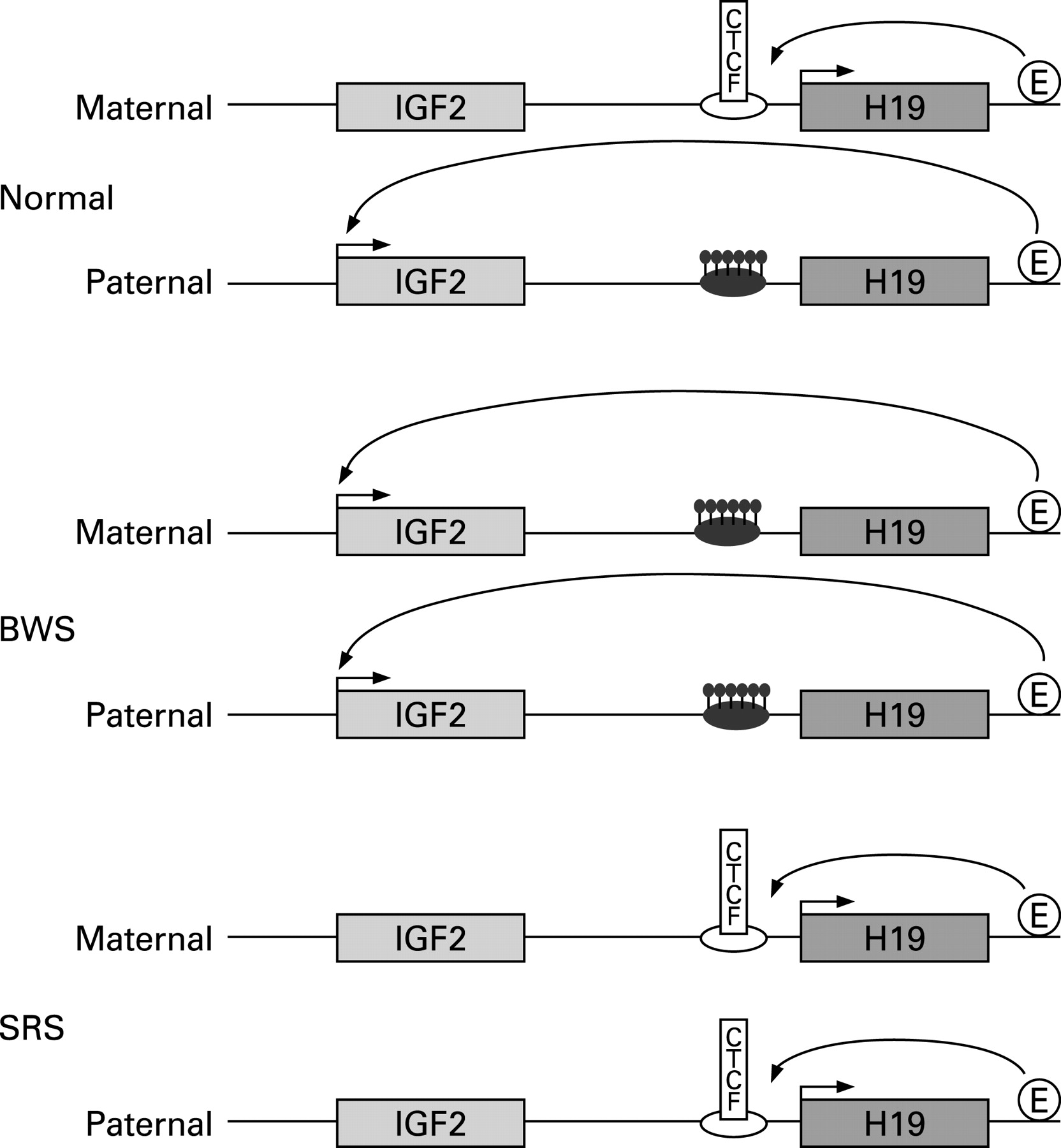
{kind=link}
Eggermann et al63 had previously described two SRS patients with maternal duplication of 11p15 material and, together with the observation of epigenetic defects in this region of BWS patients, prompted Gicquel et al53 to examine their SRS patients to see whether epigenetic defects may also explain the opposite growth phenotype. No methylation changes were seen in the centromeric ICR2 KCNQ1OT1 gene for any of their patients, but partial hypomethylation was observed in the H19 gene in lymphoblasts and fibroblasts of five SRS patients. The change was proposed to cause loss of methylation of the H19 promoter leading to biallelic H19 expression, downregulation of IGF2 and hence growth restriction. The degree of hypomethylation was correlated with the severity of the phenotype. Of critical note, one of the five SRS patients is a monozygotic twin discordant for SRS. Although her non-SRS twin showed the same partial hypomethylation in her leucocytes with concomitant biallelic expression of H19, skin fibroblast from the non-SRS twin showed no methylation abnormalities. This can be explained by the shared fetal circulation of monozygotic twins in utero. The group went on to corroborate their findings with a larger cohort of 58 SRS patients where 64% of the patients showed hypomethylation of the ICR1 region, a finding not seen in any of 68 SGA patients.64
The resulting growth restriction could be caused by over expression of either CDKN1C, a known negative regulator of cell proliferation and growth, or PHLDA2, which has been found to be upregulated in IUGR babies.65 66
Netchine et al64 also correlated the degree of hypomethylation with the severity of the growth restriction and asymmetry as well as failure to thrive, relative macrocephaly and prominent forehead. Also of interest is that the methylation is partial and variable among individuals which may give rise to tissue mosaicism. The degree and severity of body asymmetry may be a reflection of the timing of the phenomenon post-fertilisation. In addition the IGF-II serum concentrations did not appear to be altered pathologically in these individuals.64 67
It appears that the SRS patients with ICR1 methylation defects might comprise a distinct clinical cohort from the mUPD 7 patients and other idiopathic SRS cases. As early as 2001 Hannula et al68 suggested that the mUPD 7 SRS patients were phenotypically milder than others, with pre- and postnatal growth restriction, and mild or absent SRS craniofacial dysmorphology. This included slight or absent facial triangularity, absent micrognathia, absent downturned mouth corners, speech delay, poor feeding throughout childhood and excessive sweating.
The H19 methylation status of all of the patients studied to date is summarised in table 1.
A single case of SRS with maternal duplication of ICR2 has been reported recently.71 Three other patients had previously been described with maternal duplication of 11p encompassing the ICR1 and ICR2 regions. All three had IUGR but only one was considered for SRS diagnosis and did not meet the criteria.72 Eggermann et al70 have compared and contrasted the mutation types in BWS and SRS patients on chromosome 11 and have shown that the errors are of opposite parental origin including mutations, duplications and methylation deregulation.
There have been several recent reports of children born with SRS who had been conceived by assisted reproductive technology (ART).33 69 73 74 Two of these cases were found to have methylation defects.33 69 The ART embryo culture medium is a cocktail of growth factors which could conceivably have an effect on methylation of the embryo and hence its growth potential.74 Certainly studies on in vitro culture conditions on mice pre-implantation embryos have demonstrated long term physiological, neurodevelopmental and behavioural effects.75 76 Some of these changes may be due to the observed changes in mRNA expression of imprinted genes which are particularly vulnerable during the blastocyst stage.75 77 Although ART is generally considered safe, babies with low birth weight are over represented in this population of births, as well as children with BWS and Angelman syndrome (AS) caused by imprinting defects, and it is therefore of clinical significance to determine the optimal culture conditions for future ART progeny. This may be especially important when culture media contains methyl donors which may influence methylation and imprinting. There is also the consideration that ART is performed on patients with fertility problems which could carry a higher risk of passing on aberrantly imprinted genes to their offspring. Kobayashi et al78 recently described abnormal paternal methylation imprints (14.5%) and abnormal maternal imprints (20.6%) in oligospermic men.
Other chromosomes
Apart from chromosomes 7 and 11, six other chromosomes have been associated with the SRS phenotype (table 2). In some instances the patients described have had additional or atypical features.
CONCLUSION
It is now well established that 10% of SRS patients have mUPD for chromosome 7 and up to 50% have methylation defects in the imprinted domain on chromosome 11p15. This still leaves at least 40% of SRS patients with an unknown genetic aetiology. This may be explained by patient ascertainment, where many of the diagnostic criteria are shared with other syndromes. Clinicians generally agree that there are both SRS patients and SRS-like patients with a grey area between. It may be that the diagnostic criteria need to be tightened to ensure that the classical cases are identified and separated from the non-classical cases. It will be useful to make a detailed comparison of the clinical features of all SRS patients in order to identify whether the patients can be further categorised. For example, this might include those with and without limb asymmetry and to correlate genetic aetiologies with specific symptoms. For such a genetically diverse syndrome it is still conceivable that multiple genetic defects per patient could also contribute to the final phenotype.
Epigenetics is an exciting field and one which will undoubtedly prove vital in explaining many more syndromes and diseases. Various forms of cancer have for some time been linked to epigenetics, but conditions affecting neurology, behaviour and psychopathology such as schizophrenia and Alzheimer’s disease have recently also been suggested to have epigenetic components.100 101 Furthermore, the finding that diet and environment can influence epigenetic outcome, both in utero and throughout life, makes for a challenging but promising field of study.102 103 It is clear that epigenetics plays an important role in fetal growth and development but the full understanding of the mechanisms and their effect on gene function is yet to be elucidated, particularly for the benefit of clinical application. Human epigenetic models such as SRS will continue to aid us in this pursuit.
Acknowledgments
The authors wish to thank all patients and families who have participated in our research and are grateful to SPARKS, WellBeing of Women, March of Dimes, Medical Research Council and the Wellcome Trust for their generous support. SAA is a Wellbeing of Women fellow, DM is a March of Dimes fellow, and JF is a MRC, PhD student.
REFERENCES
Footnotes
Competing interests: None declared.