Article Text

Abstract
Background: Kabuki syndrome (KS) is a rare, clinically recognisable, congenital mental retardation syndrome. The aetiology of KS remains unknown.
Methods: Four carefully selected patients with KS were screened for chromosomal imbalances using array comparative genomic hybridisation at 1 Mb resolution.
Results: In one patient, a 250 kb de novo microdeletion at 20p12.1 was detected, deleting exon 5 of C20orf133. The function of this gene is unknown. In situ hybridisation with the mouse orthologue of C20orf133 showed expression mainly in brain, but also in kidney, eye, inner ear, ganglia of the peripheral nervous system and lung.
Conclusion: The de novo nature of the deletion, the expression data and the fact that C20orf133 carries a macro domain, suggesting a role for the gene in chromatin biology, make the gene a likely candidate to cause the phenotype in this patient with KS. Both the finding of different of chromosomal rearrangements in patients with KS features and the absence of C20orf133 mutations in 19 additional patients with KS suggest that KS is genetically heterogeneous.
- Appr-1”-P, ADP-ribose-1′-monophosphate
- Appr-1”-Pase, ADP-ribose-1′-monophosphatase
- BAC, bacterial artificial chromosome
- CGH, comparative genomic hybridisation
- CHARGE, coloboma, heart anomalies, choanal atresia, retardation of growth and development and genital and ear abnormalities
- CNV, copy-number variation
- DHPLC, denaturating high-performance liquid chromatography
- E, embryonic day
- FISH, fluorescence in situ hybridisation
- FLRT3, fibronectin-like domain-containing leucine-rich transmembrane protein 3
- KS, Kabuki syndrome
- NCBI, National Center for Biotechnology Information
- qPCR, quantitative PCR
- RT, reverse transcriptase
- SMART, Simple Modular Architecture Research Tool
- SNP, single nucleotide polymorphism
- TEAA, triethylamine acetate
- UCSC, University of California Santa Cruz
- Kabuki syndrome
- C20orf133
- FLRT3
- microdeletion
- array CGH
Statistics from Altmetric.com
- Appr-1”-P, ADP-ribose-1′-monophosphate
- Appr-1”-Pase, ADP-ribose-1′-monophosphatase
- BAC, bacterial artificial chromosome
- CGH, comparative genomic hybridisation
- CHARGE, coloboma, heart anomalies, choanal atresia, retardation of growth and development and genital and ear abnormalities
- CNV, copy-number variation
- DHPLC, denaturating high-performance liquid chromatography
- E, embryonic day
- FISH, fluorescence in situ hybridisation
- FLRT3, fibronectin-like domain-containing leucine-rich transmembrane protein 3
- KS, Kabuki syndrome
- NCBI, National Center for Biotechnology Information
- qPCR, quantitative PCR
- RT, reverse transcriptase
- SMART, Simple Modular Architecture Research Tool
- SNP, single nucleotide polymorphism
- TEAA, triethylamine acetate
- UCSC, University of California Santa Cruz
Kabuki syndrome (KS) or Niikawa-Kuroki syndrome is a congenital mental retardation syndrome typically characterised by postnatal growth retardation, distinct facial features with long palpebral fissures and eversion of the lateral third of the lower eyelids, typical eyebrows (reminiscent of the make-up of actors of Kabuki, a traditional Japanese theatrical performance), a broad and depressed nasal tip, prominent earlobes, persistence of fetal fingertip pads and skeletal cardiac and cleft anomalies.1–5 Major malformations of the heart, kidneys and vertebra occur frequently. Psychomotoric development is almost universally present (93%), from mild to moderately delayed and severe in some patients.6 These patients are often hypotonic and may have seizures. Endocrinological anomalies such as growth-hormone deficiency and premature thelarche are seen.
The prevalence of the syndrome is estimated to be at least 1 per 32 000 in the Japanese population,7 and is probably similar elsewhere.8 To date, no molecular cause has been determined. The sex ratio in KS is almost equal and no increased rate of consanguinity is found,9 but it is associated with advanced paternal age.5 Overall, the sporadic occurrence suggests a de novo origin of autosomal dominant mutations.7,10–14 Several people with KS features have been found to carry abnormalities of chromosomes, including the sex chromosomes and various autosomes. One patient had a pericentric inversion of the Y chromosome,7 six had a ring chromosome X or Y7,15,16 and one a 45,X karyotype.17 The reported autosomal abnormalities include an interstitial duplication of 1p13.1p22.1,18 a paracentric inversion of the short arm of chromosome 4,19 partial monosomy 6q and/or partial trisomy 12q,20 balanced translocation between 15q and 17q,21 and pseudodicentric chromosome 13.22 Given the incidence of heart defects, cleft palate and the occurrence of lower lip pits in KS, microdeletions involving chromosome 22q11 and chromosome 1q32q41 have been investigated, but no association could be detected.23,24 Bottani et al excluded mutations in the TGFβ1 and TGFβ2 genes in KS, the rationale behind this study being the resemblance between the KS and Loeys–Dietz aortic aneurysm syndrome.25 Milunsky et al reported a common 3.5 Mb duplication at 8p23.1p22 in six unrelated patients with KS phenotype, found using conventional comparative genomic hybridisation (CGH) and fluorescence in situ hybridisation (FISH).26 However, several follow-up studies using FISH or array CGH with clones covering 8p23.1p22, in a total of 112 patients with KS, could not confirm this duplication to be a common cause of KS.27–33 This multitude of reported chromosomal aberrations in KS is recognised in the variability of clinical expression of KS and its overlap with other phenotypes.
Molecular karyotyping now enables the detection of chromosomal imbalances at much higher resolution compared with conventional karyotyping. A microdeletion or microduplication would be consistent with the mostly sporadic occurrence of the “chromosomal” phenotype. In this study, we investigated four typical patients with KS using array CGH at 1 Mb resolution and identified a putative KS-causing gene in a ∼250 kb region deletion.
MATERIALS AND METHODS
Patients
Given the absence of validated diagnostic criteria for KS, all patients presented in this study were selected from a larger cohort and a consensus diagnosis was established by a group of experienced clinical geneticists. Informed consent was obtained from the patients or their legal representatives. Patient DNA and leukocyte suspensions were isolated following standard protocols. In total, 20 patients with KS were selected by clinical geneticists in Leuven (n = 4), Maastricht (n = 5) and Paris (n = 11).4,28
Patient report
The index patient with del(20)(p12.1) has been reported previously (patient 1 in Schrander-Stumpel et al8). She is the second child of healthy unrelated parents and she is currently 15 years old. She was born at 39 weeks after a delivery with forceps, with a weight of 3200 g (50th–75th centile) and length of 48 cm (25th–50th centile). A cleft palate and severe feeding problems were present. At the age of 5 months, the cleft palate was surgically closed, but the feeding problems persisted. At that time, the clinical diagnosis of KS was made. Development at 1 year of age was 8 months, according to the Bayley motor and mental scale. At 14 months, her weight and height were below third centile (6.5 kg and 69.5 cm), her occipital frontal circumferences was 44.2 cm (3rd–10th centile). Development was moderately retarded. She had a premature thelarche. At 11 years, secondary sexual development started and some fat deposition around the waist was evident (weight 31.6 kg (10th–25th centile) and height 130 cm (third centile)). Cardiac ultrasound was normal. Renal investigations revealed a left sided vesico-uretral reflux grade II and an ectopic small right kidney of 7.7 cm (located in the right hemipelvis, (SD −2.9)). She had habitual bladder and bowel disturbance. Ophthalmological findings included hypermetropia, bilateral astigmatism and alternating strabismus. Recurrent middle-ear infections needed transtymponal drains. She has short fifth fingers with clinodactyly and persistent fetal pads on the fingertips and pedes plani, a short perineum and a small urachus cyst. She is hypotonic with sialorrhoea and has striking muscle hypoplasia with relative subcutaneous fat excess, especially in the abdominal region. Her facial appearance is distinct, with long palpebral fissures and everted lower eyelids, a broad nasal tip, cleft palate, oligodontia and pre-auricular pits.
Fluorescence in situ hybridisation
FISH was performed as described previously using the P1-derived artificial chromosome clones 1043K1 and 375N15 at 8p to exclude duplications.34
Real-time quantitative PCR
Quantitative PCR (qPCR) was performed as described previously.34 Primers were designed using PrimerExpress software (Applied Biosystems, Foster City, California, USA) and analysed for the uniqueness of the sequence by using BLAT (University of California Santa Cruz (UCSC) genome browser) and BLASTN (National Center for Biotechnology Information (NCBI) database) analysis and for repeats using RepeatMasker (supplementary table 1; available online at http://jmg.bmj.com/supplemental). For the selection of the primers for the exons in the qPCR technique, we used NM001033086, NM001033087, AK125899 and the Vega transcripts of gene OTTHUMG00000031919 (this is the gene ID of C20orf133) in the Ensembl database. The qPCR was performed (Universal SYBR Green PCR Master Mix without UNG; Applied Biosystems) on an automated system (ABI7000; Applied Biosystems), in accordance with the manufacturer’s guidelines. PCR conditions were 50°C for 2 minutes, denaturation at 95°C for 10 minutes, and 40 cycles of amplification at 95°C for 15 seconds and 60°C for 1 minute. Specific PCR amplification was assessed by dissociation curve analysis. Quantification was performed using the ΔCt method and compared with the reference genes. Primers 5′-CCC-AAG-CAA-TGG-ATG-ATT-TGA-3′ and 5′-GAG-CTT-CAT-CTG-GAC-CTG-GGT-3′ in the tumour protein p53 gene, 5′-CAT-CTC-ATG-GTG-GCC-CTA-GTG-3′ and 5′-CTG-CTT-TGC-ATC-AAA-GAC-TGC-T-3′ in the annexin A3 gene (ANXA3) and 5′-TTT-TCA-TTT-TCC-TGG-CCT-TGG-3′ and 5′-TGA-CGG-CCA-GGA-GAA-GAC-AT-3′ in the olfactory receptor, family 2, subfamily B, member 2 (OR2B2) gene were used as a reference for the qPCR.
Specific clinical features in index patient with KS, phenotypic features in KS in general and expression levels of C20orf133 in different mouse embryonic tissues and stages
Paternity testing
Paternity testing was performed by co-amplification the 17 autosomal STRs vWA (12p12-pter), D2S1338, TPOX (2p25.1-pter), D3S1338, FGA (4q28), D5S818, CSF1PO (5q33.3-34), D7S820, D8S1179, TH01 (11p15.5), D13S317, Penta E (15q), D16S539, D18S51, D19S433, D21S11 and Penta D (21q) as described by Decorte et al.35,36 The index patient and both parents were analysed. A statistical analysis was performed following the procedure in Decorte et al.35,36 This analysis is based on the allele frequency in a Belgian and an American population (Penta A and Penta D) and an a priori chance of 50%.
Sequence analysis
The C20orf133 (AK131348) and the FLRT3 (a nested gene located within intron 3 of C20orf133) genes were amplified using primer sets for each exon (supplementary table 2; available online at http://jmg.bmj.com/supplemental), and PCR products were sequenced in both directions, then analysed (ABI3130; Applied Biosystems and Ensembl database, release 42).
Denaturating high-performance liquid chromatography
Denaturating high-performance liquid chromatography (DHPLC) analysis was performed according to the manufacturer’s instructions (Transgenomic Wave system; Transgenomic, Cheshire, UK) on genomic DNA of C20orf133 exon 14, using the primers 5′-TGC-ATA-TCA-CAT-TTC-TTT-TAT-TTT-TCA-3′ and 5′-CCA-CGC-ACA-CAC-ACA-GGT-AT-3′. A 10 μl aliquot of approximately 10 ng/μl crude PCR product was loaded onto a chromatography column (DNASep; Transgenomic). DNA was eluted from the column by a linear acetonitrile gradient in 0.1 mmol/l triethylamine acetate (TEAA) buffer at a constant flow rate of 1.5 ml per minute. The gradient was formed by mixing buffer A (0.1 mmol/l TEAA) and buffer B (0.1 mmol/l TEAA, 25% v/v acetonitrile). The temperature of the oven for optimal heteroduplex separation at partial DNA denaturation was deduced from melting profiles of the DNA sequence, obtained with Navigator V.1.6.2 software (Transgenomic). Elution patterns of patient samples were compared with those of normal control samples. The total run time was 2.5 minutes and gradient conditions were 52.9–62.9°C in 2 minutes. Column temperature was 56.1°C.37
Array CGH
Array CGH at 1 Mb resolution was carried out as described previously.34 The chromosome 20 tiling path clone set was derived from type RP clones, plates 1 (offsets A1, A2, B1, B2) and 2 (offsets A1, A2, B1) and from CT type clones, plate 1 (offset A1). The plates were obtained from BACPAC Resource Center (Children’s Hospital, Oakland Research Institute, Oakland, California, USA, http://bacpac.chori.org). The hybridisations and the analyses were performed as described for the 1 Mb array experiments.38 The threshold for an abnormal intensity ratio for a deletion is log2(3/2)–2SD. The latter is the standard deviation of all intensity ratios.38
In situ hybridisation
Mice were handled according to the guidelines of the Committee for Animal Experiments, University of Leuven, Belgium. In situ hybridisation on wild-type Mus musculus was performed at different stages of embryogenesis: E12.5, 14.5, 16.5 and 18.5. Embryos were isolated from pregnant mice and sacrificed by cervical dislocation. The day of plug detection was considered to be embryonic day (E)0.5. The embryos were fixed overnight in 4% paraformaldehyde in phosphate-buffered saline at 4°C and dehydrated in 70% ethanol before embedding in paraffin wax. Chromogenic in situ hybridisation on 7 μm thick paraffin sections was performed with an antisense riboprobe labelled with digoxigenin-UTP (Roche Diagnostics, Basel, Switzerland) on an automated in situ hybridisation instrument (Ventana Discovery; Ventana Medical Systems, Tucson, Arizona, USA) using commercial kits (Ribomap and Bluemap kits; Ventana Medical Systems). A fragment containing 447–1449 bp of AK134694 (NCBI database) was used as template for the cRNA probe. Pretreatment was performed with the mild CC2 procedure and hybridisation was carried out at 70°C for 8 hours. Post-hybridisation washes were performed for for 32 minutes at 70°C in 0.1× saline sodium citrate buffer and repeated three times, then the colour allowed to develop for 8 hours.
Quantitative reverse transcriptase PCR
Mouse adult brain and kidney, whole E11.5 embryos and mouse E18.5 tissues of liver, brain, lung, thymus, heart, intestine, bladder and kidney were used for RNA extraction (QIAquick RNA extraction kit; Qiagen, Valencia, California, USA), in accordance with the manufacturer’s protocol. A 4 μg aliquot of RNA was treated with DNase (Fermentas) before cDNA synthesis (SuperScript III; Invitrogen). cDNA was diluted four times for analysis. Primers were designed using PrimerExpress software (Applied Biosystems) and analysed for the uniqueness of the sequence using BLAT and BLASTN analysis and for repeats using RepeatMasker software (table 1).
Reverse transcriptase (RT)-qPCR was performed (qPCR MasterMix Plus for SYBR-Green I without UNG; Eurogentec, San Diego, California, USA) on an automated system (ABI7500; Applied Biosystems), in accordance with the manufacturer’s guidelines. PCR conditions were 50°C for 2 minutes, denaturation at 95°C for 10 minutes and 40 cycles of amplification at 95°C for 15 seconds and 60°C for 1 minute. Specific PCR amplification was assessed by dissociation curve analysis. Quantification was performed using the ΔCt method and compared with the reference genes. Primers 5′-GCA-TTT-CAA-CAG-GCA-TTT-ATG-G-3′ and 5′-TTA-ATC-GTA-CCT-AGG-GCA-ATG-AC-3′ within the mouse homologue AK134694 gene (UCSC genome browser) and the primers 5′-ACC-CAC-ACT-GTG-CCC-ATC-TAC-3′ and 5′-AGC-CAA-GTC-CAG-ACG-CAG-G-3′ in the β-actin gene were designed and used as a reference for the qPCR.
Bioinformatics
Multiple alignments of amino acid sequences was performed with ClustalW software (http://www.ebi.ac.uk/clustalw/) using the default parameter settings.39
RESULTS
FISH and 1 Mb array CGH results
In all patients, an 8p23.1 duplication, previously suggested to be causative for the KS,24 was excluded (data not shown).28,30 DNA of four patients with KS was hybridised using a bacterial artificial chromosome (BAC) array CGH at 1 Mb resolution. For three patients, the array CGH results were normal. In a fourth patient, a log2 intensity ratio of −0.83 of one clone (RP5-855L24, 14.55–14.69 Mb) at chromosome 20, band p12.1, was seen, indicating the presence of a deletion (data not shown). FISH analysis with RP5-855L24 on metaphase spreads of the fourth patient (hereafter referred to as the index patient) showed a signal on only one chromosome 20, thus confirming the presence of a deletion (fig 1A). FISH analysis with RP5-855L24 on metaphase spreads of the parents showed a signal on both chromosomes 20 (data not shown), suggesting a de novo deletion. Paternity testing confirmed the father to be the biological father (p<0.01).
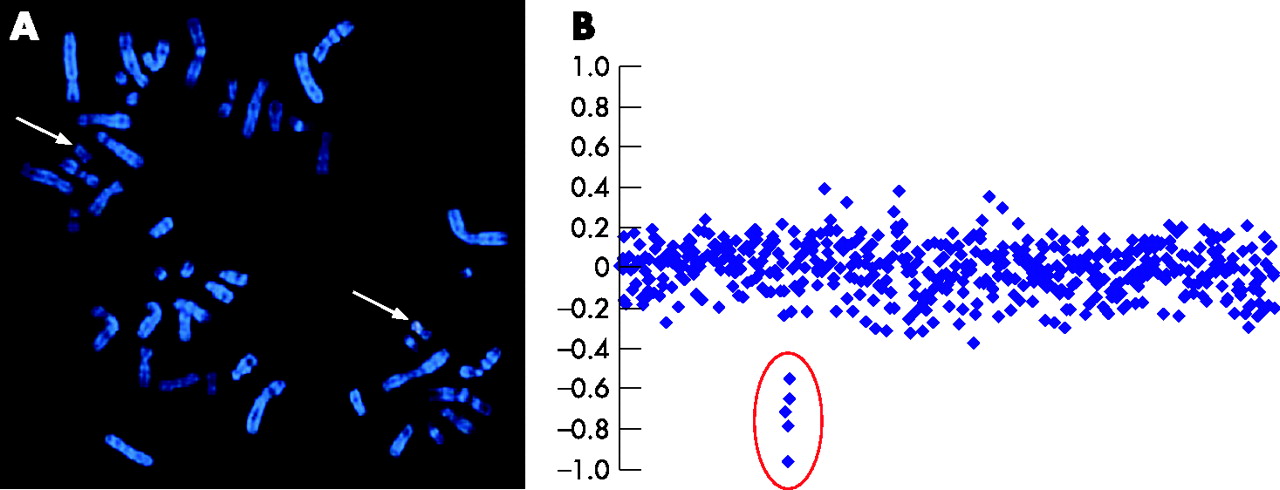
(A) Fluorescence in situ hybridisation (FISH) using Spectrum Orange-labelled RP5-855L24. Broad arrow, normal chromosome 20, thin arrow, derivative chromosome 20. (B) Full-tiling chromosome 20 array ratio profiles using DNA from a patient with a deletion and reference DNA from a normal individual. The x axis represents the clones ordered from the 20p to the 20q telomere according to their clone position in the February 2004 Ensembl freeze. The y axis marks the hybridisation ratio plotted on a log2 scale.
Fine mapping the size of the deletion
To refine the size of the deletion, we analysed the DNA of the index patient using the full-tiling chromosome 20 array CGH (fig 1B). Four BACs, namely CTD-2340K11 (14.52–14.66 Mb), CTD-2280K18 (14.53–14.68 Mb), CTD-2110N14 (14.53–14.65 Mb) and RP11-631F5 (14.63–14.78 Mb) have an average log2 intensity ratio of −0.78. The BAC RP11-318C17 (14.76–14.93 Mb) has a log2 intensity ration of −0.55, whereas RP11-582I19 (14.43–14.59 Mb) and RP11-224A21 (14.83–15.01 Mb) show normal intensity ratios. FISH using RP11-318C17 expressed a normal signal on the normal chromosome 20 and a weak signal on the second chromosome 20, indicating that this clone was partially deleted. Taken together, the deletion spans about 250 kb. Recently, large-scale benign copy-number variations (CNVs) have been detected in human populations.40 To confirm that the de novo deletion in our patient is not a benign CNV, both the database of genomic variants and the genomic variation in 270 HapMap families (Redon track, Ensembl release 42, December 2006) were investigated, and confirmed that the deletion is not a known benign variant.40,41
C20orf133 as a candidate gene for KS
The deletion in the DNA of the index patient is located within C20orf133 (AK131348 from the NITE Biological Resource Centre, or GC20P013971). This gene contains 17 exons and spans 2.06 Mb, located at 13.92–15.98 Mb. The gene structure of C20orf133 and the putative protein encoded by it is evolutionarily conserved. In particular, the N-terminal half of the putative protein is highly conserved across species. From amino acid 1–243 (of a total of 425 amino acids), the human protein shows ⩾96% similarity with the Pan troglodytes, Macaca fascicularis and M musculus orthologues, 84% similarity with Xenopus laevis and 79% with Danio rerio. This part of the protein contains an (ADP-ribose-1”-monophosphate (Appr-1”-P) processing domain, belonging to the macro domain family. The macro domain is described in approximately 300 of the proteins stored in the Simple Modular Architecture Research Tool (SMART) database. The C-terminal region of the protein is less conserved: similarity with P troglodytes is still 98%, but drops down to 76% for M fascicularis, 57% for M musculus and only 33% and 32% for the D rerio and X laevis orthologues, respectively.38 The DNA of the index patient shows, together with intronic sequences, a deletion of the entire exon 5 of this gene. This exon is located within the N-terminally conserved region (fig 2B) and encompasses 117 nucleotides, thus removing 39 amino acids from the patient’s protein but not causing a frameshift downstream (fig 2A,B). C20orf133 (Unigene cluster name Hs.570367) mRNA has been shown to be expressed in fetal and adult human brain, thymus, skeletal muscle, liver, pancreas, prostate, kidney and lung (UCSC genome browser and GenCards: Unigene Electronic Northern blotting results).
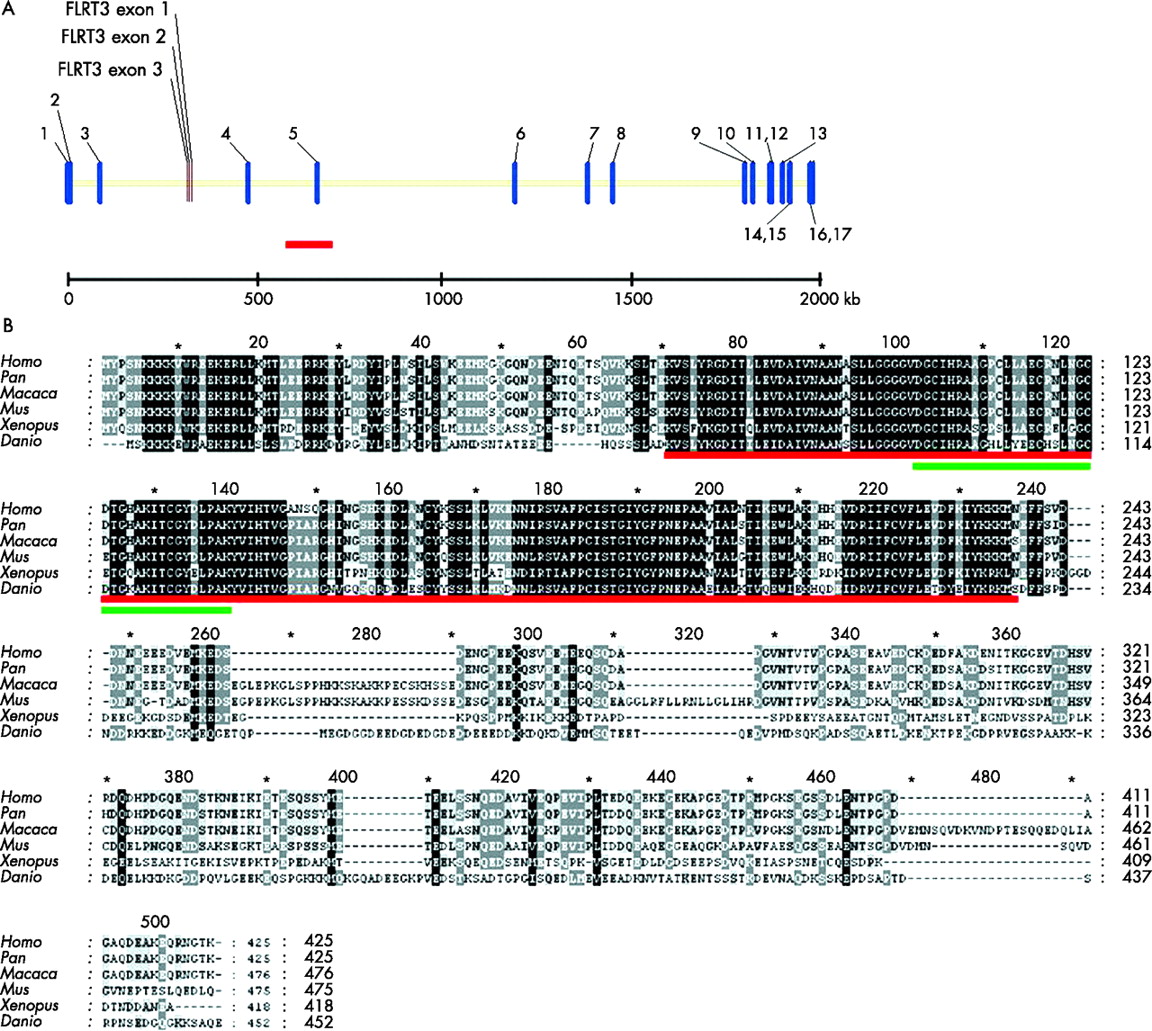
A Schematic view of the exons and introns of the gene C20orf133 and FLRT3 at a genomic level, designed using Vector NTi10. C20orf133 contains 17 exons and is 2 054 882 bp in size. FLRT3 contains three exons, is 13 621 bp in size, and is in the reverse direction to C20orf133. The red line indicates the deletion of the index patient. The scale is indicated in kbp. (B) Multiple alignment of the C20orf133 protein between Homo sapiens, Pan troglodytes (chimpanzee), Macaca fascicularis, Mus musculus, Xenopus laevis and Danio rerio. The red line indicates the macro-Appr-Pase-like domain. The green line indicates the deletion region of the index patient.
Expression pattern of C20orf133 in the mouse embryo
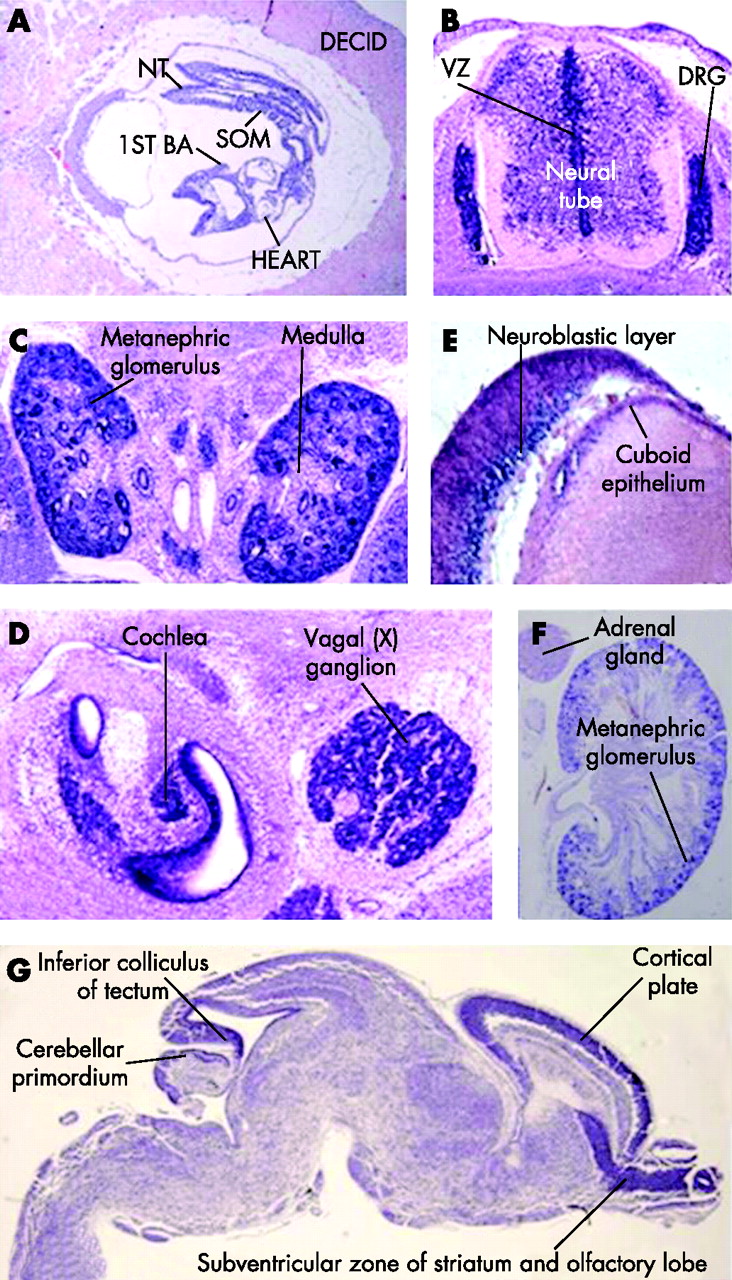
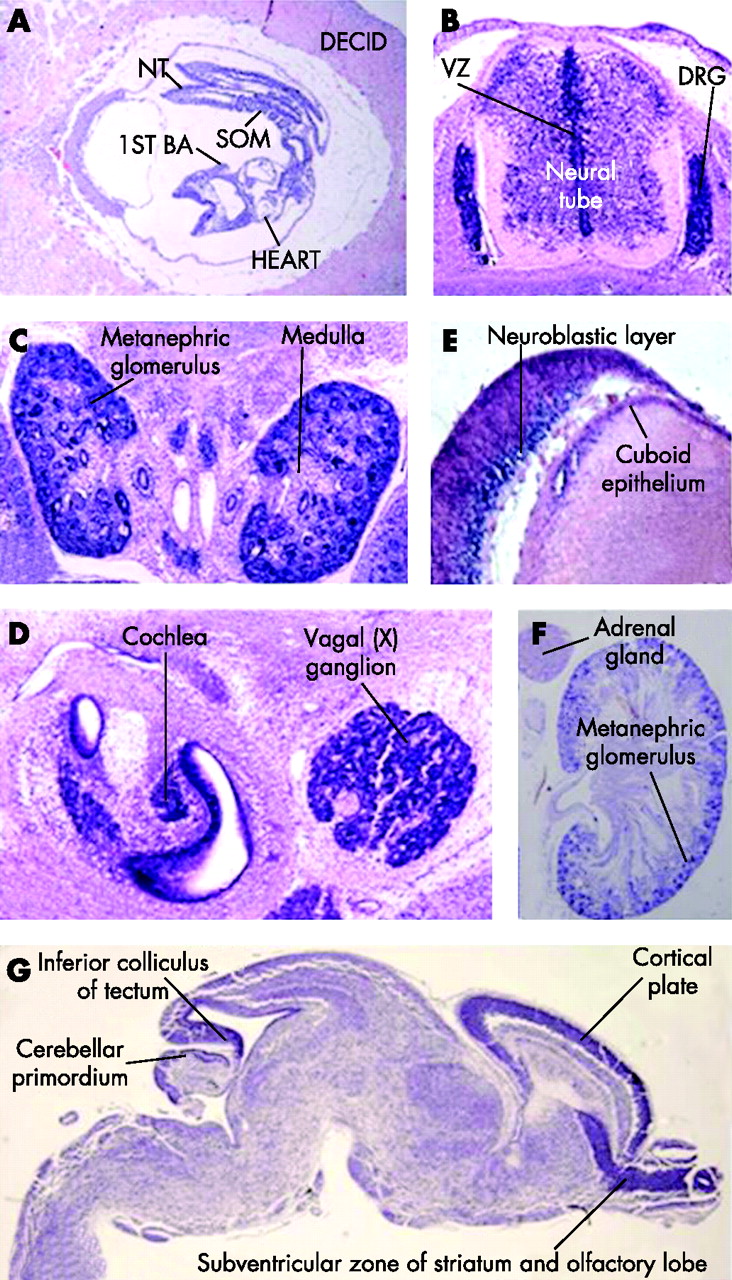
To gain insight in the relations of this gene with the patients’ phenotype, expression of the mouse orthologue of C20orf133 mRNA was studied using in situ hybridisation with an AK134694 cDNA riboprobe (NITE Biological Resource Centre; http://www.nite.go.jp) in wild-type fetuses at different stages of embryonic development. At E12.5, significant expression levels were seen in the neural tube and in the ganglia of the peripheral nervous system (the dorsal root and cranial ganglia) (fig 3A and data not shown). At developmental stage E14.5, a high level of expression was found in the metanephric glomeruli of the kidney, whereas the medullary region was devoid of mRNA (fig 3B). In addition, this gene was found to be expressed in the epithelium lining the lumen of the gut and stomach, and the seminiferous tubules, lungs, dorsal root ganglia and spinal cord (data not shown). In the cranial region, increased levels of C20orf133 expression were seen in the epithelial and mesenchymal components of the tooth-bud condensations, the epithelium of the primitive nasal cavity, in cells lining the vestibulocochlear and cochlear ducts, and the cranial ganglia (fig 3C and data not shown). In the E16.5 embryo, the lung and kidney maintained C20orf133 expression. Specific hybridisation was also detected in the papilla of the whisker follicles. In the eye, strong in situ staining was seen in the cuboid epithelium of the lens and the inner nuclear (neuroblastic) layer of the retina (fig 3D and data not shown). In addition, the brain, and in particular the ventricular zone, which is also seen in the neural tube, became positive for C20orf133 mRNA at this developmental stage (fig 3E). The expression in the brain was also clear at E18. Expression in the myocardium of the heart was seen at developmental stage E16.5 (data not shown). Just before birth, at E18.5, C20orf133 continued to be expressed in the brain, with relatively high levels of expression in discrete regions: the subventricular zone of striatum and olfactory lobe, the cortical plate, cerebellar primordium and the inferior colliculus of the tectum (fig 3G). In P0 kidney, expression of C20orf133 expression was maintained in the methanephric glomeruli, whereas it was not detectable in the adrenal gland (fig 3F).
{kind=link}
{kind=link}
{kind=link}
The embryonic expression pattern of the mouse orthologue of C20orf133 (AK134694). (A) Sagittal section of an E8.5 embryo within the deciduum (decid) reveals a widespread gene expression in all intra-embryonci and extra-embryonic tissues. The decidual tissue surrounding the embryo is largely devoid of C20orf133 mRNA, (B) Transverse section (at the level of the thorax) through an E12.5 embryo showing the spinal cord and the dorsal root ganglia (DRG) shows particularly strong expression of the mouse orthologue of C20orf133 (AK134694) in the condensed DRG (red arrowhead). Lower, but still significant expression levels can be sen in the neural tube, in particular in the ventricular zone (VZ). (C) Transverse section through E14.5 kidneys shows a strong in situ signal. (D) Cranial section at E14.5. High mRNA content is seen in cells lining the vestibulocochlear and cochlear ducts, and in the cranial ganglia. (E) In the E16.5 eye, strong in situ staining can be seen in the cuboid epithelium of the lens and the inner nuclear (neuroblastic) layer of the retina. (F) In situ staining on a kidney of a newborn (P0) mouse demonstrates significant expression of the mouse C20orf133 orthologue in the kidney, whereas the adrenal gland has no detectable expression. (G) Sagittal section through an E18.5 brain reveals expression throughout the brain, with particularly high levels of expression in the subventricular zone of the striatum and olfactory lobe, the cortical plate, cerebellar primordium and the inferior colliculus of the tectum. BA, branchial arch; decid, deciduum; nt, neural tube; som, somites.
The presence and level of expression of C20orf133 mRNA was also monitored using RT-PCR on multiple tissues and at different embryonic stages. The mRNAs of a whole E11.5 embryo, of liver, eye, brain, lung, thymus, heart, intestine, bladder and kidney at stage E18.5, and of adult kidney and brain were used. Strong expression was seen in stage E18.5 and adult brain. Low, but marked expression levels were seen in the E11.5 embryo, kidneys of embryonic stage E18.5 and adult mice, and E18.5 eyes and bladder. Expression seen in the heart at stage E18.5 was 45 times lower than in the brain at stage E18.5.
Mutation screening in other patients with KS
We investigated whether other patients with KS carried either deletions or point mutations in C20orf133. FISH analysis using RP5-855L24 as a probe on metaphase spreads of 14 of the 19 other patients with KS was performed and no deletion was detected. No metaphase spreads were available for the five patients from Maastricht. Full-tiling chromosome 20 array CGH was performed on the DNA of the five patients with KS from Maastricht and the three other patients from Leuven. There was insufficient DNA from the Paris group to perform these analyses. No imbalances were detected. To exclude other mutations in the coding sequence, all C20orf133 exons and their intron–exon boundaries in the genomic DNA of the other 19 patients with KS were sequenced. A single base-pair replacement (single nucleotide polymorphism; SNP) in exon 14 (c.19C→T) was detected in 7 patients. This SNP is not present in the Human Genome Organisation SNP database. To determine whether the SNP was a common polymorphism, DHPLC was performed on 82 control DNA samples (164 alleles). The SNP was found to be heterozygous in 25 controls, and 3 controls were homozygous for the base-pair substitution. Hence, we conclude that the SNP is a benign polymorphism. Because sequencing analysis may not detect whole exon deletions or duplications that may be small enough to be below the detection limit of the current CGH array, we performed exon-specific qPCR on the available genomic DNA of the nine patients screened with the tiling array of chromosome 20. This approach confirmed the deletion of exon 5 in the index patient, but no other deletions or duplications were detected in the other nine patients.
Mutation analysis in FLRT3
Because genomic rearrangements and thus deletions can influence the expression of nearby genes, the effect of the detected microdeletion in the index patient might be indirect, disturbing gene function(s) in the proximity. Because neither point mutations nor deletions of the C20orf133 gene were detected in the remaining patients with KS, we investigated whether FLRT3, a nested gene located within intron 3 of the C20orf133, could be affected. FLRT3 (fibronectin-like domain-containing leucine-rich transmembrane protein 3) is a transmembrane modulator of fibroblast growth factor–mitogen-activated protein kinase signalling in vertebrates and may be involved in receptor signalling protein activity, transferase activity and cell adhesion. However, neither deletions nor duplications were detected in the clones covering FLRT3 in the nine patients with KS screened using the chromosome 20 full-tiling path array. Subsequently, the three FLRT3 exons and their intron–exon boundaries were sequenced in the DNA of all 20 patients with KS, but no mutation was found.
DISCUSSION
We report a de novo microdeletion of ∼250 kb at 20p12.1 in a 15-year-old girl with the KS, using array CGH with a 1 Mb resolution. This was a fortunate finding, as the resolution of the array is lower than that of the deletion size. Several observations suggest that C20orf133 is a causative gene for the observed phenotype in the index patient. First, the deletion in this patient occurred de novo in the macro-Appr-Pase-like domain of the putative protein, thereby probably disrupting the enzymatic activity. Second, the expression pattern of the C20orf133 gene during mouse embryonic development supports its importance in the development of various tissues and organs, such as those affected in this patient. The gene contains a macro functional domain, which links it with the chromatin structure. Recently, several congenital malformation syndromes have been described as caused by haploinsufficiency of a gene involved in chromatin remodelling.42–47
In Mus musculus, both in situ hybridisation and RT-PCR showed that C20orf133 is expressed during embryonic development of the brain, in particular the ventricular zone. Just before birth, at E18.5, C20orf133 remains expressed in the brain, with relatively high levels of expression in discrete regions: the subventricular zone of the striatum and olfactory lobe, the cortical plate, the cerebellar primordium and the inferior colliculus of the tectum. The high levels of expression in the brain across a wide range of developmental stages and its persistence during adulthood is consistent with a role for the gene in mental development, and may reflect a requirement for C20orf133 in axonal outgrowth and functioning of the adult cortex. The gene is also expressed in other embryonic tissues that are typically affected in patients with KS. High levels of expression in embryonic kidney/urinary tract have been found. One study found renal malformations in 28% of tindividuals with KS,48 but such malformations might be underdiagnosed because they sometimes remain asymptomatic. In the current study, expression was seen in several craniofacial regions, which, given the specific facial characteristics of patients with KS, can be expected. C20orf133 was expressed in the mesenchymal components of the tooth-bud condensations at E14.5. Interestingly, dental anomalies such as hypodontia, malocclusion, microdontia and small dental arches, were seen in 68% of patients with KS in one study.48,49 Expression of the gene in the cells lining the vestibulocochlear and cochlear duct at stage E14.5, possibly explaining hearing loss was reported in up to 82% of patients with KS.48 In the developing mouse eye, strong hybridisation was seen in the cuboid epithelium of the lens and the inner nuclear layer of the retina, which could explain the frequency of colobomata and cataracts in KS.3 Expression in the heart is low, despite congenital heart disease being reported in 42% of patients with KS.48 Interestingly, in our index patient, no heart defect was seen after physical and ultrasound investigation. An overview of the mouse expression data, the phenotype of the index patient and the features of KS are provided in table 1.
The C20orf133 gene contains a macro domain (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/). Previously identified as displaying Appr-1”-P processing activity, the macro domain may play roles in distinct ADP-ribose pathways. In vivo evidence suggests that ADP ribosylation is an important post-translational modification that has a role in DNA repair, transcriptional activation and repression, telomere and chromatin biology, long-term memory formation, DNA binding, and DNA and/or RNA unwinding, among other processes.50 To date, no genes containing macro domains have been implicated in mental retardation and/or developmental delay. However, it is striking that several syndromes with mental retardation and multiple congenital anomalies result from haploinsufficiency of genes involved in DNA repair, transcription, chromatin biology and long-term memory formation, exactly the same processes in which macro domain-containing proteins are known to play a role. Possibly, the macro domain family of proteins may represent a novel class of dose-sensitive genes that may cause developmental disorders when mutated.
Genevieve et al reported that one of the patients with KS in their study had a clinical overlap with CHARGE syndrome.51 This syndrome was recently shown to be caused by mutations in or deletions of CHD7, a member of the chromodomain helicase DNA-binding genes, which have a unique combination of functional domains, including two N-terminal chromodomains, an SNF2-like ATPase/helicase domain and a DNA-binding domain.52 ADP–ribose binding affects the function of the Alc1 protein, a protein with homology to the Snf2 ATPase/helicase.53 Therefore, the alteration of the phosphorylation status of ADP–ribose by C20orf133 may have an influence on the activity of this Swi/Snf chromatin remodelling factor and thus explain the clinical overlap between some patients with CHARGE syndrome and KS.
Screening for mutations, deletions or duplications in 20 other patients with KS did not reveal any mutations within the C20orf133 candidate gene. Possibly, mutations outside the coding region as well as epigenetic changes might cause the KS phenotype. We also cannot exclude the presence of as yet unknown genes within the region, which might be affected by this deletion. However, it seems likely that KS is genetically heterogeneous, and that mutations in other genes may result in phenocopies. The accumulating findings of several different chromosomal rearrangements in patients with KS summarised above may pinpoint several loci that may cause KS.
Recently, other syndromes associated with multiple congenital anomalies and mental retardation have been shown to be genetically heterogeneous. For example, Noonan syndrome can be caused by mutations in the protein–tyrosine phosphatase nonreceptor-type 11 (PTPN11) gene,43 or by mutations in the V-Ki-Ras2 Kirsten rat sarcoma 2 (KRAS2) viral oncogene homologue,46 both components of the Ras pathway, or by mutations in the Son of Sevenless Drosophila homologue 1 gene (SOS1).47 The CHARGE syndrome phenotype can, in addition to mutations in the CHD7 gene, also be caused by a mutation in the semaphoring-3E gene (SEMA3E).44 Rubinstein–Taybi syndrome can be caused by mutations in the gene encoding the transcriptional coactivator CREB-binding protein (CREBBP)42 or by mutations in the E1A-binding protein 300-kDa (EP300) gene.45
In conclusion, C20orf133 is a viable candidate gene for the phenotype in the index patient. Further evaluation is needed to determine to what extent this gene could be involved in the aetiology of KS. In addition, this study further illustrates the value of array CGH to localise genes causing developmental disorders. The fortuitous finding of a microdeletion below the 1 Mb resolution of our array demonstrates that high-resolution array CGH will enable the functional identification of more genes involved in the aetiology of KS and other clinical genetic syndromes.
Electronic database information
-
NCBI accession numbers of the sequences used for alignment are NM_080676.5 (Homo sapiens), XM_001136712.1 (Pan troglodytes), AB173156 (Macaca fascicularis), CAM14292 (Mus musculus), BC060026.1 (Xenopus laevis) and NP_956843 (Danio rerio).
Acknowledgments
We wish to thank the MicroArray Facility, Flanders Interuniversity Institute for Biotechnology (VIB) for their help in the spotting of the arrays and the Mapping Core and Map Finishing groups of the Wellcome Trust Sanger Institute for the initial clone supply and verification. This work was made possible by grants OT/O2/40, GOA/2006/12 and Centre of Excellence SymBioSys (Research Council K.U.Leuven EF/05/007), Catholic University of Leuven. We thank Mrs R Thoelen for her technical support in FISH analyses, Dr E Schollen forhelp in the performance of the DHPLC, and Dr K Martens for his contribution to this article. We are also indebted to Professor D Huylebroeck for support and expert discussion.
REFERENCES
Supplementary materials
Files in this Data Supplement:
Footnotes
-
Published Online First 22 June 2007
-
Competing interests: None declared.