Article Text

Abstract
Background: The cell surface glycoprotein E-cadherin (CDH1) is a key regulator of adhesive properties in epithelial cells. Germline mutations in CDH1 are well established as the defects underlying hereditary diffuse gastric cancer (HDGC) syndrome, and an increased risk of lobular breast cancer (LBC) has been described in HDGC kindreds. However, germline CDH1 mutations have not been described in patients with LBC in non-HDGC families. This study aimed to investigate the frequency of germline CDH1 mutations in patients with LBC with early onset disease or family histories of breast cancer without DGC.
Methods: Germline DNA was analysed in 23 women with invasive lobular or mixed ductal and lobular breast cancers who had at least one close relative with breast cancer or had themselves been diagnosed before the age of 45 years, had tested negative for a germline BRCA1 or BRCA2 mutation, and reported no personal or family history of diffuse gastric cancer. The full coding sequence of CDH1 including splice junctions was amplified using PCR and screened for mutations using DHPLC and sequencing.
Results: A novel germline CDH1 truncating mutation in the extracellular portion of the protein (517insA) was identified in one woman who had LBC at the age of 42 years and a first degree relative with invasive LBC.
Conclusions: Germline CDH1 mutations can be associated with invasive LBC in the absence of diffuse gastric cancer. The finding, if confirmed, may have implications for management of individuals at risk for this breast cancer subtype. Clarification of the cancer risks in the syndrome is essential.
- lobular breast cancer
- germline mutations
- CDH1
- familial breast cancer
Statistics from Altmetric.com
The existence of a strong hereditary predisposition to breast cancer has been recognised for more than a century. Germline mutations in BRCA1 and BRCA2 have been shown to account for approximately one-third of hereditary breast cancers among young women with the disease. Mutations in other genes such as TP53, PTEN, STK11, CHEK2 and ATM account for a small proportion of hereditary breast cancer syndromes, often with distinct clinical features.1 However, in many families with breast cancer, no predisposing gene mutation can be identified. Although the existence of other strongly predisposing genes is controversial, the search for additional breast cancer susceptibility genes remains an active area of investigation.
The CDH1 (epithelial cadherin, OMIM 192090) gene is composed of 16 exons located on chromosome 16q22.1.2 The calcium-dependent molecule E-cadherin, a key regulator of cell adhesion, is the protein product of CDH1,3 and is commonly used in the immunohistochemical evaluation of breast cancers, discriminating between lobular and ductal histologies. Germline inactivating mutations in the CDH1 account for one-third of kindreds with hereditary diffuse gastric cancer (HDGC),4 defined as having ⩾2 cases of DGC in first degree relatives, with at least one documented case of DGC before the age of 50 years, or multiple cases of gastric cancer of which at least one is confirmed as DGC before the age of 50.5–7 Germline CDH1 mutations are inherited in an autosomal dominant manner, and are highly penetrant, conferring a cumulative risk of DGC of 67% in men and 83% in women.5 Recently, an excess of invasive lobular breast cancer (LBC) (including mixed ductal and lobular histology) has been reported in families with HDGC.5–8 Like DGC, LBC show histological features consistent with loss of cell–cell adhesion, and in a substantial majority, immunohistochemical technique show absence of E-cadherin.9 10 As in the case of sporadic DGC, >50% of sporadic infiltrating LBC harbour inactivating somatic CDH1 mutations accompanied by loss of heterozygosity.11 We identified probands with invasive LBC or mixed ductal breast cancer and LBC, and either early age at diagnosis or family history of breast cancer, systematically from a breast cancer databank, and analysed their peripheral lymphocyte DNA to assess possible germline mutations in the CDH1 gene.
METHODS
A group of women from the Dana Farber Cancer Institute were retrospectively identified from among a group with breast cancer who had provided signed informed consent for a banking protocol approved by the institutional review board. The consent permitted collection, storage and analysis for research of medical records, peripheral blood and tumour specimens. Participants also completed a risk factor questionnaire including unconfirmed family cancer history information stored in a linked database. Specimens were stored in the annotated Dana-Farber/Harvard Cancer Center SPORE CORE Laboratory Blood Repository, which has been maintained since 2000. The criteria for the identification of the index cases were established at the beginning of the collection and included women who had documented invasive LBC or mixed ductal breast cancer and LBC at any age, no reported relatives with gastric tumours and either (1) family history with ⩾2 cases of breast cancer in first or second degree relatives in the maternal and paternal lineage, including third degree relatives in the paternal lineage; or (2) LBC or mixed breast cancer diagnosed in the proband before 45 years of age independent of family history (table 1). Because LBCs are observed in carriers of germline BRCA1 and BRCA2 mutations, the cohort was restricted to women whose germline BRCA1/2 status was known. Those with germline mutations in the BRCA1/2 breast cancer susceptibility genes were excluded from the analysis. BRCA1/2 rearrangement analyses (BART; Myriad Genetic Laboratories, Salt Lake City, Utah, USA) had been performed clinically on only two probands; however, the prevalence of BART-detected mutations is no more than 3% in “severely affected” kindreds (R Wenstrup, Myriad Genetics, personal communication), so was not performed on our cohort.
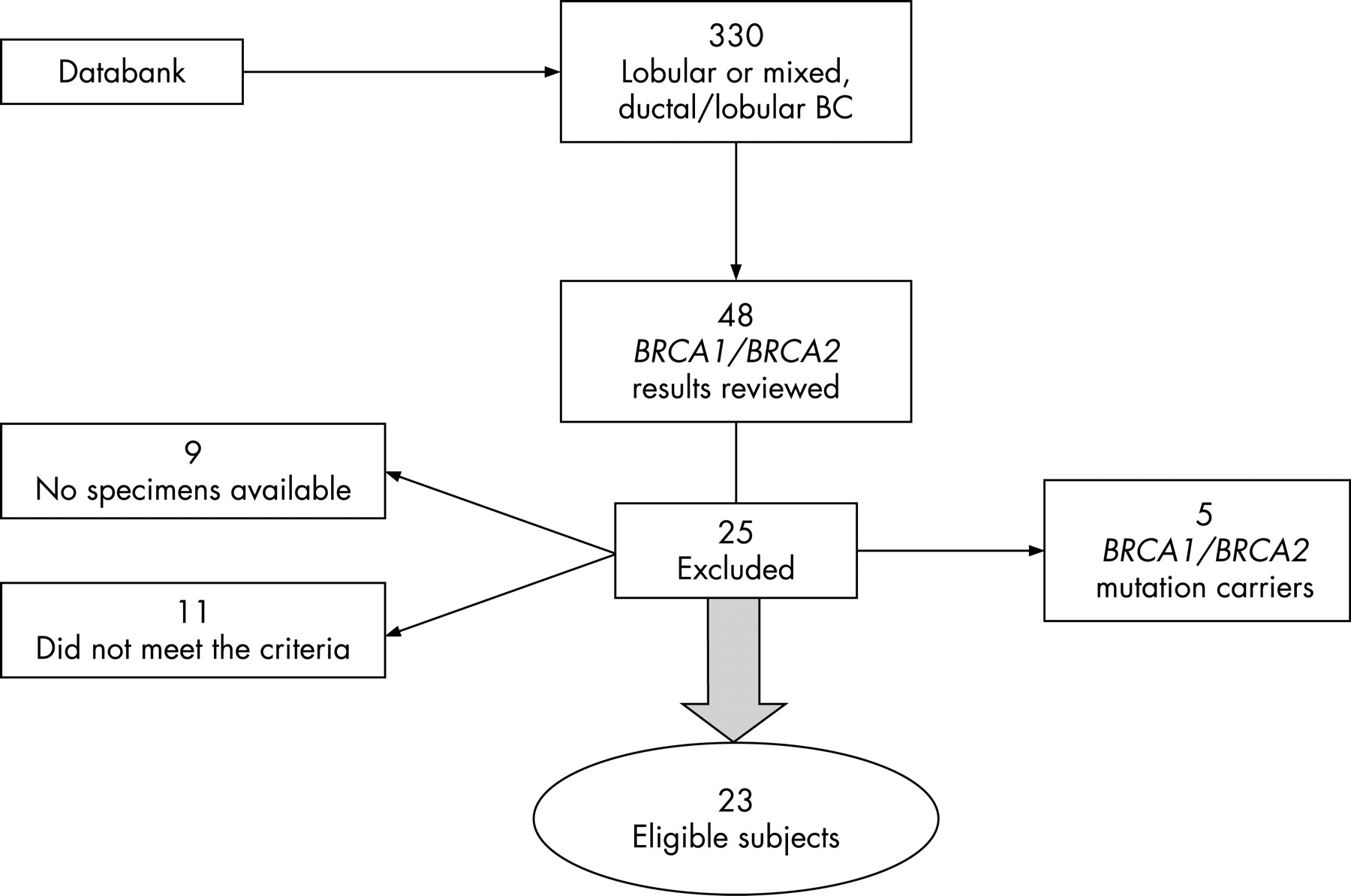
In total, 330 women with invasive LBC or mixed ductal breast cancer and LBC were identified from the databank, which has enrolled >2000 newly diagnosed breast cancer patients at Dana Farber Cancer Institute since 1999. Family history information provided by the patient at enrolment was available for >90% of cases, but could not be directly confirmed under the terms of the protocol, which precluded further patient contact. In all, 48 of these women had had DNA analysed for BRCA1 or BRCA2 germline mutations, identified in clinical testing or in the course of other research. Of these, five were excluded because of a positive BRCA1 (n = 2) or BRCA2 (n = 3) mutation identified by sequence analysis in the patient or close relatives. Of the 43 women meeting histological criteria who had tested negative for BRCA1 and BRCA2 mutations, 9 were excluded because there was no blood specimen for DNA extraction available from the core laboratory, and 11 were excluded because of failure to meet age or family history criteria. Therefore, the analysis was limited to 23 women with documented invasive LBC (9) or mixed ductal and lobular (14) breast cancers who had previously tested negative for germline BRCA1 and BRCA2 mutations (fig 1). Nineteen of these women met the first eligibility criterion, and four women met the second. The median (range) age at breast cancer diagnosis was 45 (36 to 66) years for the entire group, 46 (36 to 66) years for women meeting the first criterion and 40.5 (36 to 42) years for those in the age-related category. Medical record documentation of histopathology was assembled. Family history was confirmed with medical records where possible. All pathology slides were reviewed at time of clinical evaluation at the Brigham and Women’s Hospital. After the cohort was finalised and clinical information linked to specimens, all identifiers were removed, in accordance with protocol stipulations.

Genomic DNA was extracted from blood samples in the Dana Farber/Harvard Cancer Center Breast Cancer SPORE core laboratory at Dana Farber Cancer Institute using a Qiamp DNA Blood Midi kit (Qiagen, Valencia, California, USA). Analyses detailed below were performed at the Centre for Translational and Applied Genomics at the British Columbia Cancer Agency, Vancouver, British Columbia.
CDH1 analysis
Mutational analysis
Samples with insufficient DNA for complete mutational analysis of CDH1 underwent whole-genome amplification using the GenomiPhi DNA amplification kit (GE Healthcare Bio-Sciences Inc., Quebec, Canada) according to the manufacturer’s instructions. Briefly, 10 ng of DNA (10 ng/μl) was mixed with 9 μl of sample buffer containing random hexamer primers and heated to 95°C for 3 min. After cooling, 9 μl of reaction buffer and 1 μl of enzyme (Phi29 DNA polymerase) was added to the sample and incubated at 30°C for 18 hrs. The sample was then heated to 65°C for 10 minutes to inactivate the enzyme. Amplified DNA was purified by ethanol precipitation prior to denaturing high-performance liquid chromatography (DHPLC) analysis. The full coding sequence of CDH1 including splice junctions was amplified by PCR and screened for mutations using DHPLC. Primer sequences and conditions were as previously described.7 PCR products that had shown a potential variant with DHPLC were sequenced in both directions starting from a fresh PCR product. Before sequencing, the PCR products were purified using a gel extraction kit (Qiagen MinElute; Qiagen, Mississauga, ON). Sequencing was then performed (Big Dye Terminator V.3.1 Cycle Sequencing Kit; Applied Biosystems, Foster City, California, USA) and analysed (ABI Prism 310 Genetic Analyzer).
CDH1 promoter methylation analysis
CDH1 promoter methylation analysis was performed in microdissected tumour material from the proband. DNA was extracted using the Invisorb Spin Tissue Mini Kit (Invitek, Berlin, Germany) following manufacturer’s instructions. Approximately 200 ng of DNA were treated with an EpiTect Bisulfite Kit (Qiagen, Valencia, California, USA). Unmethylated cytosines were converted to uracil, whereas methylated ones remained unmodified. Bisulphite-treated DNA from white blood cells was methylated in vitro with M.SssI DNA MeTase and used as a positive control for methylation determination. The CDH1 promoter CpG island 3 was amplified by PCR using flanking primers (sequences available upon request) specifically designed for bisulphite-treated DNA sequences without CpG sites, and sequenced for methylation status determination.
Loss of heterozygosity analysis
Loss of heterozygosity (LOH) analysis was performed using DNA extracted from microdissected tumour and adjacent normal tissue. The CDH1 promoter common polymorphism −60C/A and the CDH1 exon 4 mutation site were used as intragenic markers for LOH analysis in DNA extracted from tumour and normal material from the proband. DNA was amplified by PCR and sequenced for each site with the aim of determining whether the wild-type allele was under-represented in tumour DNA compared with the sequencing profiles obtained from normal breast epithelia and constitutional DNA.
RESULTS
Of 23 women with documented invasive LBC or mixed ductal breast cancer and LBC who had previously tested negative for germline BRCA1 and BRCA2 mutations, one had a novel mutation in the CDH1 gene detected by DHPLC and confirmed by direct sequencing (fig 2B). Heteroduplexes that form in PCR samples having internal sequence variation display reduced column retention time relative to homoduplexes found in control samples. The 517insA mutation is located near the 5′ end of the CDH1 gene and results in a premature stop codon, eliminating all of the transmembrane and intracellular domains and the majority of the extracellular domain of the protein.

{kind=link}
{kind=link}
The mutation 517insA was found in a woman whose LBC was diagnosed at the age of 42 and whose mother reportedly had developed LBC at the age of 28. The mother’s diagnosis was confirmed by her doctor’s notes. No other breast or gastric cancers were reported in the family (fig 2A). The proband’s breast cancer was negative for E-cadherin (CDH1) by immunohistochemistry, indicating that a second molecular event, causing the complete inactivation of the CDH1 gene, had occurred (fig 2C, D). We searched for inactivation of the wild-type allele in a microdissected tumour sample from the proband. Promoter methylation analysis was performed, but no methylated alleles were found (data not shown). Subsequently, LOH analysis was performed in the same tumour sample using CDH1 distal and proximal microsatellite markers as well as intragenic markers. No loss of genetic material was found using CDH1 flanking markers (data not shown).
The LOH using intragenic markers revealed a different scenario: the sequencing of CDH1 −160C/A polymorphism showed equal peak heights for both alleles in tumour material from the proband, suggesting that LOH is not occurring at this specific 5′ end of the CDH1 gene. In contrast, the sequencing analysis for CDH1 exon 4 performed in tumour DNA shows a clear reduction of the peak heights of the wild type compared with the mutant allele. This reduction could not be observed in the sequencing analysis of constitutional DNA or in DNA from normal breast epithelia. This result is suggestive of LOH downstream of the promoter region of the gene and encompassing at least exon 4.
DISCUSSION
In this report, we describe the finding of a germline CDH1 mutation in a woman with LBC whose family history includes additional LBC but no gastric cancer. Germline mutations in CDH1 have been previously associated with marked risk of diffuse gastric cancer (67–83%), the dominant tumour in HDGC syndrome.5 Recent observations have noted an excess of invasive LBCs in some HDGC kindreds.7 8 12 The estimated cumulative lifetime risk of breast cancer in women with germline CDH1 mutations calculated for 11 DGC families is 39%.8 A penetrance analysis of four families with a founder CDH1 mutation confirmed the increased risk for breast cancer, with a cumulative risk of breast cancer of 52% (95% CI 29% to 94%).13
Previous efforts to identify germline CDH1 mutations in familial breast cancer patients have not been very informative. In a Swedish study, 19 patients with familial breast cancer whose tumours showed loss of heterozygosity at the CDH1 locus tested negative for germline CDH1 mutations.14 However, most (10 of 19) of these cases were ductal carcinomas and one was medullary, a ductal subtype. Of the remainder, two were LBC and one was mixed ductal breast cancer and LBC; information on the other five tumours was not included. As loss of E-cadherin characterises >90% of LBCs and only 5–10% of ductal histologies, this distribution of histological subtypes is unexpected.15 Lei et al did not identify a germline CDH1 mutation in 13 patients with familial LBC.16 However, in this small cohort, a positive family history was not clearly defined either for degree of relation or number of family members with breast cancer. The search for CDH1 germline mutations in a series of 65 LCIS patients also yielded negative results.17 One study has proposed that the CDH1 missense mutation 1774G→A (A592T) is a risk factor for comedo-type carcinoma, a pathological variant of ductal carcinoma in situ.14 A second germline missense mutation (1876G→A (F626V)) has been reported in an individual with LBC; no family cancer history is included in the report.18 The pathogenicity of these two missense mutations is not known.
The CDH1-encoded protein E-cadherin is a calcium-dependent cell–cell adhesion glycoprotein comprised of an extracellular domain, a transmembrane region that bridges the plasma membrane and a highly conserved cytoplasmic tail.2 19 It is one of the key molecules for the establishment of the intercellular junction complex and for the adhesive properties between epithelial cells. The cytoplasmic domain of E-cadherin directs the β-catenin-mediated interaction with the actin cytoskeleton and p120 controls the strength of cell–cell adhesion by regulating cadherin stability and retention at the cell surface. It acts in a zipper-like fashion at the tight junctions of adjacent epithelial cells.19 Downregulation of CDH1 leads to the disruption of the tissue architecture and the increase of invasive properties of the malignant cells of epithelial origin.20 The loss of CDH1 expression can occur as a result of various genetic mechanisms. For example, in sporadic DGC, somatic mutations preferentially target exons 7 and 9, and promoter hypermethylations account for biallelic silencing of CDH1 expression in >50% of this type of tumour,10 21 whereas in most sporadic LBC, complete silencing of CDH1 is achieved by mutations scattered along the gene accompanied by either CDH1 promotor methylation or LOH.10 22
The 517insA mutation described in this report is located near the 5′ end of the CDH1 gene. LOH at the mutation site was found in the analysis of two separate samples extracted from the proband’s LBC. In contrast to gastric cancers from germline CDH1 mutation carriers, in which promoter methylation is the commonet second hit,21 23 in this case of LBC we found that the second hit is through LOH. Interestingly, in the current study, LOH was not identified using CDH1-flanking LOH markers, but using polymorphic intragenic markers, namely the mutation site in exon 4, which revealed an intragenic deletion that encompasses at least exon 4 of the CDH1 gene. This mechanism was previously reported in a tumour from a HDGC CDH1 mutation carrier.21 23
Our finding suggests that genetic heterogeneity may also characterise familial invasive LBC. LBC comprise 9% of the breast cancer in carriers of germline BRCA2 mutations and only 3% in mutation carriers of BRCA1.24 The present report demonstrates that CDH1 germline mutations occur in 4.3% (1/23) of LBC probands. Therefore, if these results are confirmed in larger series, CDH1 testing may become part of the evaluation of women with LBC in whom features suggesting the presence of hereditary predisposition are present. In other known cancer syndromes, histopathological information defines subsets of cancers linked to particular genes. For example, medullary thyroid cancer is associated with activating germline mutations in the RET proto-oncogene, but follicular thyroid cancer occurs excessively in Cowden syndrome with germline PTEN mutations. Clear-cell renal carcinoma is the commonest finding observed in the Von Hippel Lindau syndrome, whereas papillary renal-cell carcinoma is associated with germline mutations in the proto-oncogene c-MET.25 Among breast cancers, medullary, atypical medullary and basal-like tumours are more commonly observed in individuals with germline BRCA1 mutations.26 27 If confirmed, the association between invasive LBC and germline CDH1 mutations may help to guide the genetic evaluation of affected individuals and families.
The finding raises questions for the clinical management of CDH1 carriers. Further study will be necessary to determine more clearly the penetrance of germline CDH1 mutations and the proper management of women with such mutations. Although LBC represent only 8–14% of all breast cancers,28 they account for a disproportionate number of breast cancers undetectable by screening mammography. The role of breast MRI has not been defined in this cohort. Challenges already exist in the management of the DGC risk inherent in previously identified CDH1 mutation carriers. We have shown that several detection methods have low sensitivity for detecting early gastric cancer in HDGC patients, including endoscopy, endoscopic ultrasound, chromoendoscopy, and PET scanning, which failed to detect early DGC in all six patients 1 week before prophylactic gastrectomy.29 30
In summary, we report a novel germline CDH1 mutation in a woman with LBC and family history of LBC in the absence of DGC. Additional research can now focus on reliable estimates of the mutation frequency, spectrum, penetrance and range of malignancies associated with germline CDH1 mutations. Further work to identify appropriate and effective surveillance and prevention strategies for individuals at hereditary risk of LBC with and potentially without risk of DGC will also be critical.
Key points
Germline CDH1 mutation was found in a woman with lobular breast cancer and family history of breast cancer but not diffuse gastric cancer.
Loss of heterozygosity was shown in a tumour specimen from the mutation carrier.
These results, if confirmed, have implications for the genetic basis of lobular breast cancer and for the identification and management of individuals at risk.
Acknowledgments
This research was funded in part by the BC Yukon Breast Cancer Foundation to David Huntsman and by the Breast Cancer Research Foundation (BCRF) to Judy E Garber. Serena Masciari is supported by a Charles A. King Trust, Bank of America Fellowship, Co-Trustee (Boston, MA) and The Humane Society of the Commonwealth of Massachusetts Postdoctoral Research Fellowship. Nina Larsson was supported for this work by a Patterson Fellowship.
REFERENCES
Footnotes
The first two authors contributed equally to this work.
Competing interests: None declared.
- Abbreviations:
- BART
- BRCA1/2 rearrangement analyses
- DHPLC
- enaturing high-performance liquid chromatography
- HDGC
- hereditary diffuse gastric cancer
- LBC
- lobular breast cancer
- LOH
- loss of heterozygosity
- OMIM
- Online Mendelian Inheritance in Man