Article Text

Abstract
The trace metal copper is essential for a variety of biological processes, but extremely toxic when present in excessive amounts. Therefore, concentrations of this metal in the body are kept under tight control. Central regulators of cellular copper metabolism are the copper-transporting P-type ATPases ATP7A and ATP7B. Mutations in ATP7A or ATP7B disrupt the homeostatic copper balance, resulting in copper deficiency (Menkes disease) or copper overload (Wilson disease), respectively. ATP7A and ATP7B exert their functions in copper transport through a variety of interdependent mechanisms and regulatory events, including their catalytic ATPase activity, copper-induced trafficking, post-translational modifications and protein–protein interactions. This paper reviews the extensive efforts that have been undertaken over the past few years to dissect and characterise these mechanisms, and how these are affected in Menkes and Wilson disease. As both disorders are characterised by an extensive clinical heterogeneity, we will discus how the underlying genetic defects correlate with the molecular functions of ATP7A and ATP7B and with the clinical expression of these disorders.
- copper
- Menkes disease
- ATP7A
- Wilson disease
- ATP7B
Statistics from Altmetric.com
Many trace elements require a delicate homeostatic balance to ensure that the needs for normal cellular processes are met, but at the same time, toxicity due to excessive accumulation of these elements needs to be prevented. Copper is an excellent example of such a trace element. It is required for numerous cellular processes, including mitochondrial respiration, antioxidant defence, neurotransmitter synthesis, connective tissue formation, pigmentation, peptide amidation and iron metabolism (table 1).1 However, in amounts that exceed cellular needs, copper is highly toxic, owing to its potential to facilitate the production of reactive oxygen species by means of Fenton chemistry.2 Refined mechanisms have evolved to regulate intake, excretion and the cellular distribution of copper (Box 1). The importance of these regulatory mechanisms is underlined by several hereditary human disorders of copper homeostasis. These disorders can broadly be divided into two classes; (1) diseases associated with copper deficiency (Menkes disease (MD), OMIM 309400;3 and occipital horn syndrome (OHS), OMIM 304150), and (2) diseases associated with copper excess (Wilson disease (WD), OMIM 277900;4 Indian childhood cirrhosis (ICC), OMIM 215600;5 endemic Tyrolean infantile cirrhosis (ETIC), OMIM 215600;6 and idiopathic copper toxicosis (ICT); OMIM 2156007 8). The clinical expression of several of these disorders is highly heterogeneous. In this review, we discuss both the genetics and molecular–functional defects underlying MD and WD, the genotype–phenotype correlations of these disorders and how such correlations might be explained by the molecular–functional defects. Additional information of interest to the reader, but not essential to the scope of this review, is provided in the text boxes.
Copper-transporting ATPases and human disease
Diseases associated with copper deficiency
MD is an X-linked recessive disorder characterised by a general copper deficiency.3 9 The incidence of the disease is estimated to range between 1:40 000 and 1:350 000.10–12 Clinical features of MD are a direct consequence of dysfunction of several copper-dependent enzymes (cuproenzymes; table 1), secondary to an inability to load these enzymes with copper. Based on the symptoms, two forms of MD have been described; classic MD and mild MD, a less severe form. The clinical features of classic MD typically comprise neurological defects (severe mental retardation, neurodegeneration, seizures), growth retardation, hypothermia, laxity of skin and joints, hypopigmentation, and peculiar “kinky” or “steely” hair.13–15 Patients present at 2–3 months of age, and owing to the severity of the disorder, death usually occurs by 3 years of age. Patients with mild MD have a longer lifespan, and in these patients the neurological defects in particular are less profound.16–18 OHS (also known as X-linked cutis laxa, or Ehlers–Danlos syndrome type IX) is allelic to MD and its symptoms generally overlap, with the most notable exception being that neurological abnormalities are far less severe or even absent in OHS. Patients with OHS present with slightly subnormal intelligence and seizures are generally absent. Connective tissue abnormalities represent the predominant features of OHS. Characteristic is the formation of occipital exostoses resulting from calcification of the trapezius and sternocleidomastoid muscles at their attachments to the occipital bone.13 19 Treatment of MD consists of copper replacement therapy in which the following fundamental issues should be taken into consideration: (1) the block in intestinal absorption of copper must be bypassed, (2) patients must be identified and treatment started as early in life as possible, (3) circulating copper must be delivered to the brain, and (4) copper must be available within cells for cuproenzyme biosynthesis.20 21 The only currently available treatment option consists of administration of copper–histidine, a naturally occurring copper–amino acid complex in serum.22 23 Although copper replacement therapy with copper histidine results in significant improvement in some patients, leading to an increased lifespan, it has not been uniformly beneficial and the prognosis of patients with classic MD remains inevitably poor. The age at which treatment is started and the severity of the disease seem to be among the main determinants for the outcome of this treatment.20 24
Both MD and OHS are caused by mutations in the ATP7A gene,18 25–27 which encodes a highly conserved copper translocating P(1B)-type ATPase with orthologues present in eukaryotes, prokaryotes and archaea.28 ATP7A mRNA is expressed in a wide range of human tissues, but its expression is notably low and sometimes even undetectable in the adult liver.25 27 The ATP7A polypeptide contains several conserved domains required for ATPase function and copper binding (Box 2). It has been estimated that one-third of MD cases arise from de novo mutations.13 Over 200 MD-causing mutations have been identified, among which small deletions/insertions, nonsense mutations, missense mutations, and splice site mutations are represented with equal frequency.29 30 Small deletions/insertions and nonsense mutations are found throughout the whole gene. In contrast, missense mutations are almost exclusively distributed between the first transmembrane domain and the stop codon. The relative lack of MD-causing or OHS-causing missense mutations within the six metal-binding sites (MBSs) located in the amino terminal tail suggests that functional redundancy exists among these six MBSs. Splice-site mutations are mostly clustered between exons 6 and 8, which encode a region just upstream of the first transmembrane domain, and between exons 21 and 22, a region encoding the last transmembrane domain. Interestingly, splice site mutations appear to be over-represented in patients with OHS.29 30
Several mouse models for MD and OHS have been described, collectively known as mottled mice.31 The mottled mouse phenotypes show a similar variability, as seen between MD and OHS patients. The dappled subtype displays the most severe phenotype; affected mice usually die during prenatal development. In contrast, blotchy mice display a milder phenotype, reminiscent of OHS. Both dappled and blotchy, as well as other mottled subtypes, are caused by mutations in Atp7a, the murine orthologue of ATP7A.32–42 Transgenic expression of ATP7A in mottled mice of the brindled subtype rescues the phenotype and partially restores the copper balance.43 Calamity, a zebrafish mutant defective in the orthologue of ATP7A (atp7a), has recently been bred.44 Calamity zebrafish display a general copper deficiency phenotype and form an excellent model to study the function of atp7a in the developing zebrafish.
Diseases associated with copper excess
WD is an autosomal recessive disorder characterised by defective copper excretion, with an estimated incidence between 1:30 000 and 1:100 000.45 Clinical features of WD result from toxic accumulation of copper, primarily in the liver and the brain, and therefore may include hepatic abnormalities (cirrhosis and chronic hepatitis, culminating in progressive liver failure), neurological defects (parkinsonian features, seizures), and psychiatric symptoms (personality changes, depression, psychosis).45 46
In some severe cases, patients present with fulminant liver failure. A characteristic feature often found in patients with WD is the Kayser–Fleischer ring, a deposition of copper in the Descemet membrane visible as a gold-brown ring around the periphery of the cornea. In addition, serum levels of the cuproenzyme caeruloplasmin are often found greatly reduced in patients with WD as a result of rapid degradation of the copper-free form of caeruloplasmin formed in patients with WD.47 48 This type of caeruloplasmin deficiency is distinct from acaeruloplasminaemia, a disorder of iron metabolism caused by mutations in the caeruloplasmin gene.49 The presentation of WD is strongly heterogeneous, even among patients with the same mutations. Differences have been found in age at presentation, severity of the disease and the predominance of hepatic versus neurological symptoms.48 Treatment of WD focuses on two aspects: (1) copper excretion from the body must be promoted, and (2) copper absorption from the diet must be reduced.45 The first aspect is best accomplished by copper chelation therapy using penicillamine,50 trientine50 or ammonium tetrathiomolybdate.51 52 The latter remains under clinical trial, but seems particularly promising as, when taken with meals, this compound also prevents absorption of copper from the diet. Dietary copper absorption is also efficiently inhibited by zinc ingestion and by omitting copper-rich dietary components. Controversy exists as to which approach constitutes the more effective treatment regimen, and proper randomised control studies to clarify this issue are lacking. In some cases, the preferred method includes initial copper chelation followed by zinc therapy to prevent remission of high copper concentration in the liver. In general, these treatment options are very effective; however, if treatment is ineffective, or in circumstances of fulminant liver failure, liver transplantation provides an effective cure.
The gene mutated in WD, ATP7B, encodes a copper-transporting P-type ATPase that is highly homologous to ATP7A.53–56 The expression pattern of ATP7B is strikingly different to that of ATP7A, as ATP7B is most abundantly expressed in the liver. Other tissues that express ATP7B include kidney, brain, lung, heart, mammary gland and placenta.53 56–58 Almost 300 mutations in ATP7B that are associated with WD development have been described.59 In contrast to MD, relatively few small insertions/deletions and splice site mutations have been identified in patients with WD; in fact, almost 60% of all identified WD-causing mutations are missense mutations. The distribution of these mutations over ATP7B is similar to the distribution of MD-causing missense mutations in ATP7A.30 Although most WD-causing mutations are rare and only reported in single families, some are more common and account for a large portion of WD cases. The most prevalent mutations are H1069Q in Europe and North America, and R778L in southeast Asia.60
ICC, ETIC and ICT form a second class of copper-overload disorders that are distinct from WD.61 62 Most patients with ICC, ETIC and ICT die at an early age due to liver failure as a consequence of decompensated liver cirrhosis. Neurological defects are not found in ICC, ETIC or ICT. Phenotypic expression of ICC, ETIC and some cases of ICT appears to be associated with both an excessive copper intake and an underlying genetic defect.5 6 8 63 Attempts to identify the genetic causes for these disorders have remained unsuccessful, although several candidate genes, including ATP7B, have been excluded.61 62
Several animal models of copper overload diseases have been described.64 Long–Evans cinnamon (LEC) rats and toxic-milk mice suffer from abnormalities in the Atp7b gene, making these valid spontaneous models for WD.65–70 In addition, an engineered Atp7b knockout mouse had recently been generated.71 The hepatic abnormalities seen in both the LEC rat and the toxic-milk mouse closely resemble those found in WD, but neurological defects have only been found in the Atp7b knockout mouse, but not in the LEC rat nor in the toxic-milk mouse.64 71 Another interesting animal model characterised by hepatic copper overload is copper toxicosis (CT) in Bedlington terriers .72 Pathophysiologically, CT is similar to WD, although neurological defects have not been found and serum caeruloplasmin concentrations are normal. Furthermore, linkage analysis has excluded Atp7b as a candidate gene for CT, suggesting that CT is more likely to be a model for ICC, ETIC or ICT.73 74 Using positional cloning approaches, a deletion in COMMD1 (formerly MURR1), resulting in complete absence of the COMMD1 protein, was identified as a genetic cause for CT.75–77 This observation initially led to the suggestion of COMMD1 as a candidate gene for human copper overload disorders. However, no disease-causing mutations in COMMD1 have been detected in several cohorts of patients with WD, ICC, ETIC or ICT61 78–82 Possibly, other genetic causes for CT in Bedlington terriers exist, as no mutation in COMMD1 could be detected in several pedigrees with affected dogs.83 84 Nine homologues of COMMD1 have recently been reported, which should be prioritised in the search for alternative genetic causes of CT.85
Functions of copper-transporting ATPases
The physiological functions of ATP7A and ATP7B can largely be deduced from the observed phenotypes in MD and WD, respectively. In MD, copper transfer across the mucosal barrier is impaired, whereas in WD hepatic excretion of copper into the bile is reduced, suggesting that both proteins are rate limiting for cellular copper export. Biochemical and clinical observations in patients with MD or WD suggested an additional role for ATP7A and ATP7B in cuproenzyme biosynthesis, as markedly reduced copper incorporation of several cuproenzymes has been found in MD and WD.47 86–90
Regulation of cellular copper export
Direct evidence for a copper export function of ATP7A and ATP7B came from cell culture studies showing that absence of ATP7A in MD-derived patient fibroblasts resulted in cellular copper accumulation and increased copper retention, despite normal copper uptake rates.91 This copper retention phenotype could be corrected by expression of either ATP7A or ATP7B.92 Consistent with these observations, overexpression of ATP7A or ATP7B in a variety of cell lines results in a decrease in copper accumulation and retention, and in an increased tolerance to elevated environmental copper levels.93–95 Direct proof for the actual copper translocation function of these proteins came from studies showing that overexpression of ATP7A or ATP7B resulted in increased translocation of 64Cu into isolated membrane vesicles, which has also been shown using purified ATP7A reconstituted in soybean asolectin liposomes.96–98 Taken together, these data suggest that both ATP7A and ATP7B function as ATP-dependent copper export pumps. The difference in phenotype between MD and WD is mainly determined by differences in tissue distribution and cellular localisation of ATP7A and ATP7B, as discussed below. Unfortunately, the effects of MD-causing and WD-causing mutations on the copper transport activity of ATP7A and ATP7B have only been determined using complementation assays of ΔCcc2 yeast. Although these yeast studies have clearly shown the inability of mutated ATP7A and ATP7B proteins to mediate copper delivery to the secretory pathway, no studies have yet been performed to directly elucidate the effects of disease-associated mutations on the transport activity of ATP7A and ATP7B. Significant efforts are currently being made to develop several new tools that allow detection of changes in intracellular copper concentrations and bioavailability.99–102 We have developed an MRE–luciferase reporter, which is a sensitive and robust tool to assess the ability of ATP7A and ATP7B to exort copper from the cytosol, and appears useful to assess the effects of MD-causing and WD-causing mutations on cytosolic bioavailable copper (van den Berghe and Klomp, unpublished observations).
Regulation of cuproenzyme biosynthesis
Loss of function of several cuproenzymes is a characteristic feature of both MD and WD. Using fibroblasts isolated from patients with MD or mottled mice, it has been shown that ATP7A deficiency directly results in reduced activities of the copper-dependent enzymes lysyl oxidase, tyrosinase, cytochrome C oxidase, extracellular superoxide dismutase and peptidylglycine α-amidating mono-oxygenase.86–90 Although not consistently found among all patients, WD is often associated with a dramatic reduction in serum caeruloplasmin levels as a result of rapid degradation of the copper-free form of caeruloplasmin secreted from the hepatocytes of patients with WD.47 48
In yeast, the orthologue of ATP7A and ATP7B, Ccc2p, serves a similar function. Ccc2p is required for incorporation of copper into the caeruloplasmin homologue Fet3p, a ferrireductase enabling growth of yeast under iron-deficient conditions.103 Incorporation of copper into Fet3p takes places within the Golgi apparatus, where Ccc2p resides under basal culturing conditions.104 Expression of both ATP7A and ATP7B rescues the growth phenotype of Ccc2 knockout yeast on iron-deficient media, making this an excellent model to study copper-transporting, ATPase-dependent cuproenzyme biosynthesis.105 106 Using this Ccc2 knockout complementation assay, effects of WD-causing and MD-causing mutations on translocation of copper into the trans-Golgi network (TGN) lumen and subsequent incorporation of copper in cuproenzymes can be determined. This approach has been undertaken by several investigators, and indicates that multiple MD-causing and WD-causing mutations diminish or even completely abrogate the ability of ATP7A or ATP7B to rescue Fet3p biosynthesis or the Ccc2 knockout growth phenotype (tables 2 and 3). Taken together, these observations imply that ATP7A and ATP7B are critical for both proper delivery of copper to the TGN and subsequent cuproenzyme biosynthesis, and that mutations in ATP7A and ATP7B that perturb this process account for many of the clinical symptoms of MD and WD (table 1).
Molecular mechanisms of ATP7A and ATP7B function and the effects of disease-causing mutations
ATP7A and ATP7B exert their functions in copper transport through a variety of interdependent mechanisms and regulatory events. These include catalytic ATPase activity, copper-dependent trafficking, post-translational modifications and protein–protein interactions. In recent years, extensive efforts have been undertaken to dissect and characterise these mechanisms, which will be discussed in this section in the context of the pathophysiology of MD and WD. As explained below, these mechanisms are highly interdependent, and specific MD-causing or WD-causing mutations can exert effects on multiple levels. Therefore, care should be taken in the interpretation of how such effects relate to the pathogenesis of MD and WD.
ATPase catalytic cycle
The ion translocation cycle by P-type ATPases is believed to occur through a general cycling model involving several discrete stages in which ATP hydrolysis drives translocation of the target ion (schematically depicted in fig 1).107 These stages are: (i) binding of the target ion, (ii) binding of ATP to the nucleotide-binding (N)-domain, (iii) ATP hydrolysis and phosphorylation of the phosphorylation (P)-domain, (iv) translocation of the target ion, and (v) dephosphorylation of the P-domain by the actuator (A)-domain.

This model suggests that copper plays a key regulatory role in the catalytic cycle of ATP7A and ATP7B. ATP7A and ATP7B contain a number of putative copper-binding sites, of which the amino terminal MBSs are the best characterised.108 109 The amino terminal tail containing these MBSs interacts with the N-domain of ATP7B, leading to inhibition of ATP binding.110 This interaction is inhibited by copper, providing a potential mechanism for copper-regulated availability of the N-domain to bind ATP.110 Consistent with this hypothesis, both ATP hydrolysis and formation of the acylphosphate intermediate (step (3) in the model) are also dependent on copper.98 111–114 The effect of copper on acylphosphate intermediate formation is cooperative, suggesting that the six amino terminal MBSs have a regulatory role in the formation of the acylphosphate intermediate.98 112 113 Indeed, MBSs 5 and 6 are required for the cooperative effect of copper on acylphosphate intermediate formation.112 However, mutation of all six amino terminal MBSs only mildly affects the rate and extent of catalytic phosphorylation of ATP7A, raising the possibility that other copper binding sites, such as the conserved intramembranous CPC motif, play an important role in the regulation of this process.111
The next steps in the model predict that copper translocation by ATP7A and ATP7B is dependent on ATP binding and hydrolysis. Indeed, translocation of copper into membrane vesicles isolated from rat liver, and in vitro copper translocation activities of ATP7A and ATP7B, are ATP-dependent.96–98 115 116 Using purified recombinant protein fragments, ATP binding to both the N-domain and P-domain of ATP7B has been found.110 117 118 Molecular modelling analysis supports this experimental observation.119 The exact binding site for ATP in the N-domain has not yet been identified, but analysis of the solution structure of the N-domain of ATP7B has implicated a number of residues in this process, including H1069, G1099, G1101, I1102, G1149 and N1150.118 Interestingly, H1069, G1099, G1101 and I1102 are all residues found mutated in WD, and H1069Q represents the most frequent WD-causing mutation in Europe and North America.59 Consistent with a role for this residue in ATP binding, the H1069Q mutation results in almost complete absence of ATP binding to ATP7B.117 However, within the N-domain, >40 different WD-causing mutations have been reported, indicating that more residues might be involved in either direct ATP binding or coordination.59 This possibility is supported by experiments showing that the WD-causing E1064A mutation results in complete absence of ATP binding to the N-domain of ATP7B.117
Molecular modelling analysis suggests that after initial ATP binding to the N-domain of ATP7B, conformational changes take place that bring the ATP binding site within the N-domain in close proximity to the P-domain.119 This could potentially promote ATP binding and phosphorylation of the P-domain, the third step in the catalytic cycle model.119 ATP binding in the P-domain of ATP7B has been suggested to take place in the vicinity of D1027.119 This residue is part of the DKTG motif, which is highly conserved in all P-type ATPases, and is presumed to be the target of phosphorylation by the γ-phosphate of ATP.107 Mutation of this aspartic acid residue in either ATP7A or ATP7B completely prevents formation of an acylphosphate intermediate, thereby supporting this hypothesis.111 113 114 Furthermore, this mutation in ATP7A is associated with a complete loss of copper translocation activity, the next step in the general model.111
Although copper translocation can be measured using isolated membrane vesicles from cells expressing ATP7A or ATP7B, or using purified ATP7A or ATP7B reconstituted in soybean asolectin liposomes, studies have yet to be performed to characterise the effects of MD-causing and WD-causing mutations on this particular step in the cycling model.96 98 Furthermore, no studies have yet been performed to unravel the role of the intramembranous copper-binding CPC motif, which is presumed to play an essential role in this step. However, it has been shown that intact MBSs in the amino terminal region of ATP7A are required for proper translocation of 64Cu in isolated membrane vesicles.120
Dephosphorylation of the aspartic acid residue is mediated by intrinsic phosphatase activity, in which the A-domain plays a key role. The signals that induce this phosphatase activity, and the mechanisms behind it, have not yet been elucidated, but an essential role has been ascribed to the conserved TGE motif. Mutation of this motif in ATP7A results in hyperphosphorylation of the protein.114 Although the TGE motif per se has not been reported to be mutated in MD or WD, adjacent residues in ATP7A and ATP7B are known sites of MD-causing and WD-causing mutations.59 114 In fact, the MD-causing L873R mutation, two amino acids upstream of the TGE motif, results in hyperphosphorylation of ATP7A.114
Copper-dependent localisation of ATP7A and ATP7B
Under basal conditions, ATP7A and ATP7B are localised within the TGN (fig 2).105 121 122 This localisation is consistent with their function in cuproenzyme biosynthesis, as several cuproenzymes are synthesised within the secretory pathway. Some controversy exists about the localisation of ATP7B, as it has also been suggested that this protein resides in an endosomal compartment.123 124 In addition, a smaller isoform of ATP7B exists, which has been localised to mitochondria.125 However, the current general agreement is that ATP7B is localised to the TGN, as this has been confirmed by several independent groups, including at ultrastructural resolution in human liver biopsies.105 126–133

A key mechanism in the regulation of the copper export function of ATP7A and ATP7B became apparent from studies showing that the subcellular distribution of both proteins was sensitive to the concentration of copper to which the cell was exposed (fig 2, Box 3). In response to raised copper levels, ATP7A reversibly relocalises to a peripheral vesicular compartment and to the plasma membrane.122 134 In polarised cells, and in intestinal tissue sections, ATP7A specifically localises towards the basolateral membrane upon copper exposure, consistent with its function in transferring copper across the intestinal barrier.135–138 ATP7A overexpressed in mouse liver tissue also localises at the hepatocyte basolateral membrane.139 Specific targeting of ATP7A to the basolateral membrane appears to be mediated through a putative PDZ binding motif present in the carboxy terminal tail of ATP7A. Deletion of this motif results in targeting of ATP7A to the apical membrane in response to raised copper levels.136 ATP7B undergoes a similar copper-induced relocalisation to a peripheral vesicular compartment. Although ATP7B has not been unequivocally detected at the plasma membrane of polarised cells, the idea that the transporter rapidly recycles between the peripheral vesicular compartment and the plasma membrane cannot be excluded.105 140 In polarised hepatocytic cell lines, exposure to high copper concentrations results in localisation of ATP7B in the proximity of the apical vacuoles, a structure reminiscent of the bile canaliculus.46 128 130 133 140 This observation is consistent with its proposed function in excretion of copper from the hepatocyte via the bile. Small amounts of ATP7B localised in the proximity of the bile canaliculus have also been detected in human liver tissues.130 Signals mediating the specific targeting of ATP7B towards the apical region seem to be present within the first 63 amino acids, a region that is not present in ATP7A.133 Taken together, these data indicate that differences in the trafficking destinations of ATP7A and ATP7B are not caused by general cell-type specific differences in regulation of polarised membrane protein localisation, but rather are determined by intrinsic signals present in their amino acid sequences. The difference in the directionality of copper-induced relocalisation of ATP7A and ATP7B illustrates why two distinct, but very similar, copper export proteins are required to ensure proper copper uptake and excretion in higher organisms. In addition, this difference, resulting in distinct physiological functions of these homologous proteins in maintaining whole-body copper homeostasis, explains the opposed copper transport defects seen in MD and WD.
Several MD-causing and WD-causing mutations are associated with defects in copper-induced relocalisation of ATP7A and ATP7B respectively, suggesting that this is a key event that precedes cellular copper excretion by ATP7A and ATP7B. Therefore, such defects might possibly be associated with the pathogenesis of MD and WD. In general, MD-causing and WD-causing mutations can result in three types of localisation defects (tables 2 and 3, fig 3). The first type displays a normal steady-state localisation of the protein within the TGN, but responsiveness to copper is lost.114 127 141 142 One mutation of interest that has been shown to result in such a defect is the MD-causing C1000R mutation in ATP7A, which disrupts the putative copper-binding CPC motif, suggesting that in this case impaired copper binding causes the lack of copper-induced trafficking to the cell periphery.114 This type of localisation defect could potentially explain some of the biochemical symptoms seen in patients with MD or WD. For example, although the G943S mutation in ATP7B prohibits copper-induced trafficking of ATP7B, this mutation still permits cuproenzyme biosynthesis in the ΔCcc2 complementation assay.127 143 Although this correlation does not exist for all mutations causing this type of localisation defect, it could explain the normal caeruloplasmin production found in some patients with WD. Unfortunately, serum caeruloplasmin levels have only been described for one patient who was compound heterozygous for the G943S mutation, and this was indeed found to be within normal range.127 Although this type of localisation defect might still permit cuproenzyme biosynthesis in MD, resulting in alleviation of the clinical phenotype, this might not always be obvious in patients, owing to an impairment in copper translocation across the mucosal barrier. More likely, this type of localisation defect could allow for the beneficial effects of copper replacement therapy in these patients. The second type of localisation defects is on the opposite side of the spectrum. Several MD-causing and WD-causing mutations result in constitutive peripheral localisation of ATP7A or ATP7B, respectively.114 126 141 Of interest is the MD-causing L873R mutation in ATP7A, targeting a residue located adjacent to the conserved TGE motif. In this case, a constitutively peripheral localisation of ATP7A as a result of this mutation has been associated with hyperphosphorylation of ATP7A, suggesting that copper-induced relocalisation is dependent on the ATPase catalytic cycle (further discussed in Box 3).114 The third type of localisation defect is probably the most common and clinically important. MD-causing and WD-causing mutations result in mislocalisation, or possibly retention, of ATP7A and ATP7B within the endoplasmic reticulum.126 127 140 144–149 Strikingly, this defect has been found for the two most common WD-causing mutations, H1069Q and R778L.126 127 148 Some controversy exists about the localisation of the H1069Q variant, as this has also been reported to localise to aggresomes.144 ER mislocalisation of proteins is often due to misfolding and associated with proteasomal degradation.150 A well-known example of this process in human disease development is the ER-associated degradation of the ΔF508 CFTR mutant in cystic fibrosis.151 In analogy, the H1069Q mutation in ATP7B indeed results in an increased proteolysis rate.144 148 Whether the increased proteolysis resulting from the H1069Q mutation is due to defects in the conformation of ATP7B remains unclear, but the solution structure of the N-domain of ATP7B containing the H1069Q mutation shows no folding defects.118

Post-translational modifications
Most efforts in investigating putative post-translational modifications of ATP7A and ATP7B have focused on the formation of the acylphosphate intermediate during the catalytic cycle. However, recent studies have led authors to suggest that both ATP7A and ATP7B are subject to basal and copper-induced phosphorylation that is distinct from the formation of the acylphosphate intermediate.152 153 Both basal and copper-induced phosphorylation could still be observed with a catalytically inactive mutant of ATP7B, indicating that these types of phosphorylation are indeed distinct from the formation of the acylphosphate intermediate, and that specific kinases are required.154 Copper-induced phosphorylation of ATP7B is partially inhibited by an inhibitor of casein kinase II.152 Phosphoamino acid analysis indicates that both ATP7A and ATP7B are phosphorylated on serine residues.153 155 In addition to phosphorylation, modification of ATP7A with N-linked glycan chains has also been found.121 Although little is known about the functional effects of ATP7A glycosylation, it is interesting that this type of modification is probably specific to ATP7A, as the amino acid sequence of ATP7B does not contain any consensus glycosylation sites at relevant extracytoplasmic domains.
Taken together, studies on post-translational modifications of ATP7A and ATP7B are still in their initial phases, but could shed important new light on our understanding of the regulation of these copper-transporting ATPases. The importance of these modifications is emphasised by the absence of copper-induced phosphorylation of ATP7B due to the WD-causing G591D mutation, and absence of ATP7A glycosylation, due to the MD-causing G1019D mutation (tables 1, 2).146 152
Protein–protein interactions
Protein–protein interactions form an essential mechanism through which many proteins exert their functions. Mapping of the protein–protein interactome provides a valuable framework for elucidating the functional organisation of the human proteome, and consequently to understanding of the molecular pathology of human disease.156 In recent years, significant process has been made in unravelling the ATP7A and ATP7B interactome (Box 4). In the following, we elaborate on two of the interacting partners of ATP7A and ATP7B that can be directly linked to the development of copper homeostasis disorders.
Owing to its toxic potential, concentrations of free copper inside the cell are extremely low. In fact, it has been estimated that yeast maintains a concentration of <1 free copper ion per cell.157 As a consequence of such low free copper concentrations, donor proteins are required for the delivery of substrate copper to copper-transporting ATPases. Originally isolated as a suppressor of oxidative toxicity in yeast, Atx1p (and its human orthologue ATOX1) was shown to be required for copper-transporting ATPase-mediated cuproenzyme biosynthesis158–160 Subsequently, it was demonstrated that ATOX1 interacts with both ATP7A and ATP7B.161 162 This interaction is conserved in yeast and bacteria, illustrating its importance.163–166 An MxCxxC-containing MBS, homologous to those in ATP7A and ATP7B, is present in ATOX1, through which it has been shown to bind copper.163 167 The interaction of ATOX1 with ATP7A or ATP7B is copper-dependent and requires intact MBSs of both ATOX1 and ATP7A or ATP7B.161–163 168–171 These data suggest that ATOX1 delivers copper to ATP7A and ATP7B, which in fact has been demonstrated in vitro.172–175 Atox1 knockout mice display a phenotype similar to that seen in patients with MD including symptoms such as growth failure, skin laxity, hypopigmentation and seizures.160 Consistent with a copper excretion defect caused by defective copper delivery to ATP7A, cultured fibroblasts isolated from Atox1 knockout mice exhibit an increase in copper retention and content.160 176 A time-dependent and dose-dependent impairment of copper-induced relocalisation of ATP7A has also been found in these cells.176 One possible defect underlying the development of MD and WD could thus be an impairment of the interaction of ATOX1 with ATP7A or ATP7B respectively. Indeed, several mutations in the amino terminal tail of ATP7B prevent its interaction with ATOX1.161 Two of these mutations, G85V and G591D, affect highly conserved glycine residues in the proximity of the MxCxxC core sequence in MBS 1 and 6 respectively, indicating an important role for this conserved residue in coordination of the ATOX1–ATP7B interaction.161 However, these mutations also result in mislocalisation of ATP7B to the ER, which might also underlie the loss of interaction of ATP7B with ATOX1 caused by these mutations.149
Recent studies have shown that COMMD1, the protein defective in CT, interacts with ATP7B and that this interaction is mediated by the amino terminal tail of ATP7B.177 Transient knock-down of COMMD1 in HEK293 cells results in increased cellular copper levels.178 These data suggest that COMMD1 and ATP7B cooperate in the excretion of copper from the hepatocyte, and that absence of the interaction between these proteins underlies the pathophysiology of CT in Bedlington terriers with the COMMD1 deletion. Strikingly, COMMD1 also interacts with ATP7A, indicating that COMMD1 has a role in general copper homeostasis that is not restricted to the liver (de Bie et al., unpublished observation). COMMD1 has also recently been implicated in several other cellular processes, including the nuclear factor (NF)-κB and HIF1 signalling pathways.85 179–184 In these two pathways, COMMD1 is thought to exert its regulatory role by regulating the proteasomal degradation of key components of these pathways.179 181 182 185 With this in mind, COMMD1 might also regulate the proteasomal degradation of ATP7A and ATP7B, thus regulating copper homeostasis. Other examples exist in which cuproenzymes or copper transport proteins, such as the copper chaperone for superoxide dismutase 1 (CCS) and hephaestin, are regulated by means of proteasomal degradation in response to altering copper levels.186 187 The WD-causing mutations G85V, L492S, G591D, and A604P result in increased binding of ATP7B to COMMD1, suggesting that disruption of this interaction could underlie the development of WD in some cases.149 The G85V and G591D mutations have also been associated with ER mislocalisation and increased degradation of ATP7B, supporting the hypothesis that COMMD1 facilitates the degradation of ATP7B.149 Recently, 10 homologues of COMMD1 that are characterised by a conserved domain have been described, and are also involved in regulation of NF-κB signalling.85 184 It would be interesting to investigate if these homologues of COMMD1 also have a functional role in copper homeostasis, possibly through interactions with ATP7B or ATP7A.
Genotype–phenotype correlations in the development of MD and WD
Proper characterisation of the defects in MD and WD is required to resolve the clinical heterogeneity that is seen in both disorders. Either environmental or genetic variations might underlie this clinical heterogeneity. For example, development of ICC, ETIC and ICT is associated with a high copper intake.5 6 8 63 Although such correlation has not been found for WD, it is possible that high copper intake might exacerbate the symptoms, whereas dietary components having low copper but high zinc concentrations might have a beneficial effect. Genetic variations in genes other than ATP7A and ATP7B might modulate the clinical expression of MD and WD. In various other disorders with a mendelian mode of inheritance, it has been shown that modifier genes can modulate penetrance, dominance modification, expressivity and pleiotrophy.188 This is exemplified by genetic modification of the severity and expressivity of cystic fibrosis by polymorphisms in various genes, including mannose-binding lectin and transforming growth factor β (reviewed by Cutting189 and Boyle190). COMMD1 has been proposed as a modifier gene for the clinical presentation of WD, as heterozygosity for a silent missense mutation in COMMD1 was found to be possibly associated with an earlier onset of the disorder in patients with known ATP7B mutations.80 In other cohorts of patients with WD, no association between variations in COMMD1 and the clinical expression of WD was found.78 81 82 However, these cohorts were not classified to include patients with WD with identical mutations in ATP7B, which should be performed to reliably assess the possible role of COMMD1 as a modifier gene for the clinical presentation of WD. Variations genes coding for prion protein and apolipoprotein E have been proposed to modify the presentation of WD, although for the latter this has not been consistently found.191–194
Notwithstanding the role modifier genes and environmental factors play, it is evident from the functional data discussed above that different mutations in ATP7A or ATP7B can potentially result in different functional effects. Therefore, it appears likely that also the type of mutation, or the residue(s) affected, modulate the clinical expression of MD or WD. For example, functional redundancy among the six amino terminal MBSs explains the relatively low amount of MD-causing or WD-causing missense mutations within the amino terminal tails of ATP7A and ATP7B. In addition, it has recently been found that translation reinitiation after a deletion in the amino terminal coding region of the ATP7A transcript produces a truncated protein containing only the fifth and sixth MBSs. Interestingly, this truncated protein was still partially functional, and the patient in which this phenomenon was found displayed a remarkably mild MD phenotype.195 A suggestive correlation between the severity of the phenotype and the extent to which missense mutations impair the function of Atp7a was recently found in three types of Mottled mice.196
Many studies have focused on establishing genotype–phenotype correlations in both MD and WD. Severe mutations such as nonsense and frameshift mutations causing insertions/deletions are generally believed to completely disrupt protein function, and would therefore be expected to yield a more severe clinical phenotype. Indeed, for WD it has been shown that severe mutations lead to an earlier onset of the disease, but a correlation with the type of presentation (neurological vs. hepatic) has not been found.197–200 Similarly, it has been suggested that severe mutations in ATP7A result in classic MD, whereas mild MD is caused by mutations that still permit residual activity of ATP7A.201 On the other hand, OHS is often associated with splice site mutations. Although these mutations are predicted to dramatically affect ATP7A mRNA splicing, expression of the normal transcript is often still detectable, indicating that minor amounts of normally spliced ATP7A expressed in these patients are sufficient to allow for an alleviation of the phenotype.18 40 202–204 In addition, it was suggested in a recent case report that, in two brothers with MD carrying the same mutation in ATP7A, differences in severity of the disease correlated with the amount of ATP7A expressed.205
Genotype–phenotype correlation analyses for missense mutations are less clear. Approximately 30% of all cases with MD result from de novo mutations, as a result of which mutations are often very rare, thus prohibiting proper genotype–phenotype correlation analysis using large numbers of patients. Unfortunately, such studies in WD are also hampered by the large number of mutations detected; most mutations are very rare, and therefore most patients with WD are compound heterozygous. In a large number of studies, no genotype–phenotype correlation could be found. As most of these studies relied on small patient cohorts, combining data from several independent studies in a meta-analysis could be useful to shed more clarity on this issue, as has been the case for the common H1069Q mutation.206 In several independent cohorts, a correlation between ATP7B H1069Q homozygosity and a neurological presentation of WD has been found.206–211 In addition, this genotype has been associated with a significantly later age at onset.198 199 206 208–215 In several other studies, one or both of these observations could not be statistically confirmed.198 207 212–220 However, a meta-analysis of all genetic studies before 2004 devoted to H1069Q genotype and phenotype indicated that overall, H1069Q homozygosity is indeed associated with a late neurological presentation of WD.206 A similar correlation with a later age at onset in WD has been found for patients homozygous for the R969Q mutation.199 In some cohorts, it seems that homozygosity for the R778L mutation correlates with an early, hepatic presentation of WD.221 222 It has been suggested that alternative splicing of ATP7B in the brain, resulting in absence of exon 8 (harbouring the R778 residue), underlies this suggested correlation.222 223 However, here also, studies could not confirm the correlation of the homozygous R778L genotype with hepatic presentation, although a tendency towards this observation was sometimes present.224–226
CONCLUSION
Mutations in the structurally and functionally highly homologous copper-transporting ATPases ATP7A and ATP7B underlie both copper deficiency and overload diseases. Tremendous progress has been made in the characterisation of the function of these proteins, and how this is impaired in MD and WD. From the data presented in tables 2 and 3, it is evident that different mutations in ATP7A or ATP7B result in a variety of defects in the molecular functions of these proteins. Consistent with this notion, to some extent a correlation of the genotype with the heterogeneous phenotypes has been found in these disorders. However, it is also very likely that some of the different mutations in ATP7A or ATP7B result in overlapping functional impairments. This overlap could form a potential bias in genotype–phenotype correlation studies, in which correlations might be missed due to such an overlap in test and control patients. To overcome this potential bias, genotype–phenotype correlations should be performed using patient groups classified primarily using the functional effects of the disease-causing mutations. This approach would also permit the performance of correlation studies with relatively mild or rare, mutations. At present, this approach is unfortunately not yet possible, as only a limited number of MD-causing or WD-associated variants of ATP7A and ATP7B have been functionally characterised (tables 2 and 3). Furthermore, a thorough characterisation of all molecular mechanisms that participate in the copper transport functions of ATP7A and ATP7B would be required, as these mechanisms are highly interdependent (as schematically suggested in fig 3). Such characterisation of mutations in ATP7A and ATP7B should be an important focus of future studies on the functional genetics of MD and WD, and will result in valuable insights into the molecular pathogenesis of MD and WD.
Box 1 Copper homeostasis
Copper is taken up from the diet in enterocytes and subsequently effluxed to the liver through the portal vein. This step is blocked in MD and OHS (blue bar in fig 4). From the liver, copper is distributed to the general circulation to provide tissues with required copper. Excretion of the majority (98%) of copper from the body is mediated by biliary export, indicating that the liver plays a central role in the regulation of body copper homeostasis.227 Copper excretion from the liver is defective in WD, ICC, ETIC and ICT (red bars in fig 4), resulting in accumulation of excess copper in the liver. To ensure copper homeostasis on the cellular level, refined mechanisms to regulate copper uptake, distribution and excretion have evolved. Copper import in the cell takes place via the copper transporters 1 and 2.102 228 229 After uptake, distribution of intracellular copper is facilitated by a group of proteins called copper chaperones, which function to deliver free copper to its sites of utilisation. Several human copper chaperones have been described; CCS delivers copper to SOD1 in the cytosol and mitochondria,230 231 COX17 delivers copper to the cytochrome C oxidase complex in mitochondria,232 233 and ATOX1 delivers copper to the copper-transporting ATPases ATP7A and ATP7B in the trans-Golgi complex.158 159 These copper-transporting ATPases play an essential role in the export of copper from the cell. Dysfunction of these proteins underlies the development of MD, OHS and WD, and forms the topic of this review.

Box 2 Structure of copper-transporting ATPases
ATP7A and ATP7B are highly homologous and both belong to the heavy metal-transporting P(1B)-type ATPases. The basic topology of ATP7A and ATP7B is depicted in figure 5. The polypeptide sequences of ATP7A and ATP7B are 54% identical, however two sequence inserts are present in ATP7A that are not present in ATP7B (the position of these sequences are marked with an arrow). Several conserved motifs are present in both ATP7A and ATP7B that are characteristic for the P-type ATPase protein family.107 These motifs are required for ATP catalysis and include the nucleotide binding domain (N-domain; depicted in blue), the phosphorylation domain (P-domain; depicted in green) and the actuator domain (A-domain; depicted in red). Highly conserved signature residues are present in these motifs; SEHPL in the N-domain, DKTG in the P-domain, and TGE in the A-domain. The specific functions of these domains are discussed in the text. Within the amino terminal tail, six MBSs; depicted in purple) are present, each containing the core sequence MxCxxC. These MBSs bind Cu(I) in a stoichiometry of one atom of Cu(I) per MBS.108 109 Structural analysis of separate MBSs of ATP7A or ATP7B has shown a conserved βαββαβ folding structure.234–240 The sequence and structure of these MBSs are highly conserved in evolution and to other proteins, such as the copper chaperone ATOX1.28 These amino-terminal MBSs in ATP7A and ATP7B are required for several aspects of their function, including copper translocation, incorporation of copper in cuproenzymes, ATPase activity, localisation and trafficking, and protein–protein interactions.106 111 112 114 120 131 136 162 168 171 241–243 Strikingly, however, bacterial and yeast orthologues of ATP7A and ATP7B only contain one or two MBSs, indicating that the six MBSs in ATP7A and ATP7B might be partially redundant. In general, it is believed that P-type ATPases contain binding sites for their substrate ion within their transmembrane regions to facilitate transfer of these ions across the membrane barrier.107 Trans-membrane domain 6 contains a highly conserved CPC motif (depicted in purple) that is characteristic for heavy metal transporting P-type ATPases.244 245 Peptides containing this motif have been shown to bind copper, indicating that the CPC motif could indeed be involved in the actual transfer of copper across the membrane.240
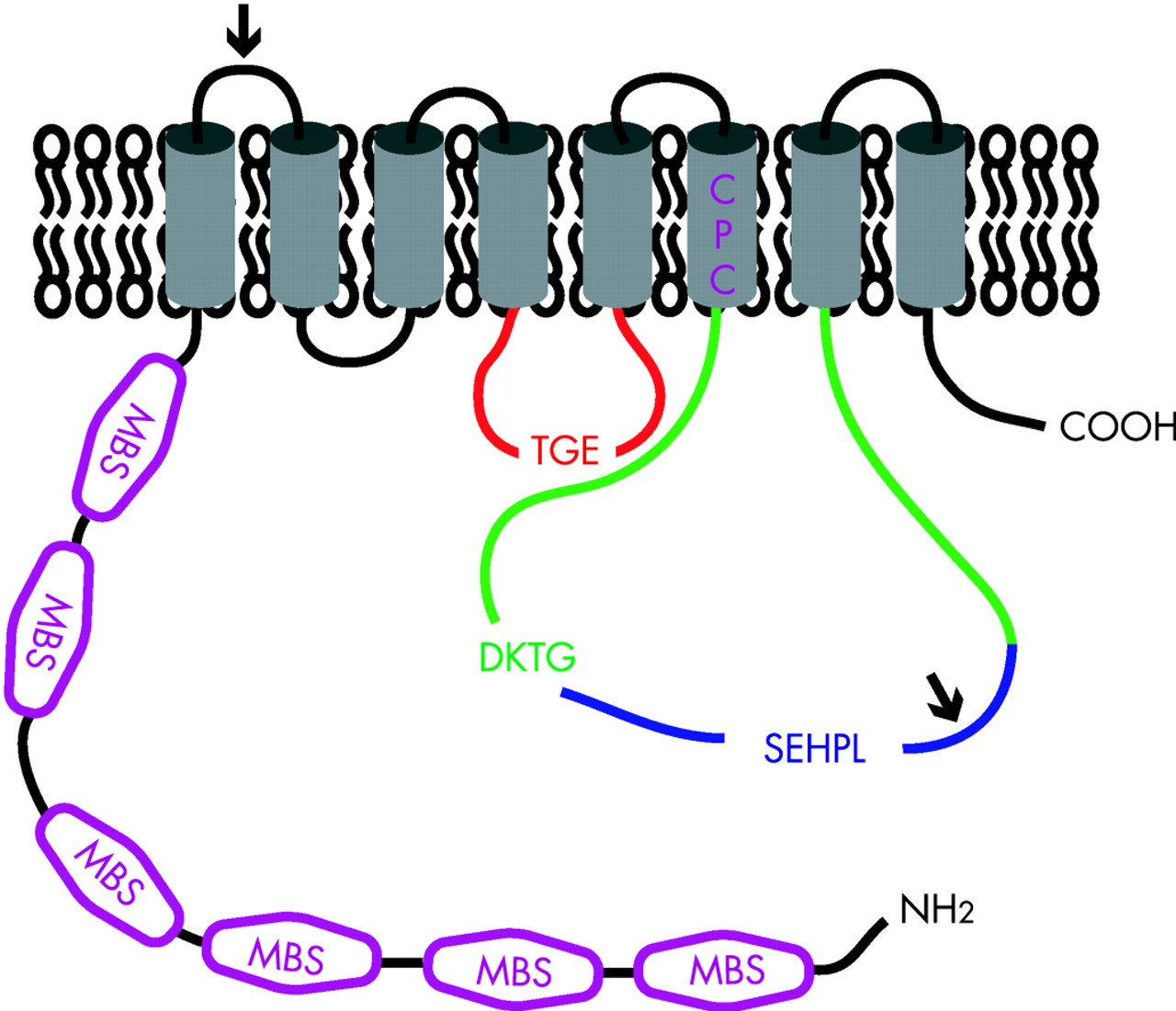
Box 3 Signals and mechanisms mediating copper-induced trafficking of ATP7A and ATP7B
Signals and mechanisms underlying the localisation and copper-induced trafficking of ATP7A and ATP7B have been the focus of many studies. Steady-state TGN localisation of ATP7A has been shown to be mediated by a putative TGN-targeting signal within transmembrane domain 3.147 Although transmembrane region 3 of ATP7B shows a high degree of conservation with that of ATP7A, it is unknown if it serves the same TGN-targeting function. Copper-induced relocalisation of ATP7A and ATP7B appears to be initiated by direct binding of copper to ATP7A and ATP7B. Deletion or mutation of all amino terminal MBS or the putative copper-binding CPC sequence results in an inability to induce the copper-dependent relocalisation.114 131 136 242 243 Strikingly, however, the presence of only one intact amino terminal MBS is sufficient for normal copper-induced relocalisation.131 242 243 246 247 This observation indicates that the six different MBSs have redundant functions, at least in copper-induced relocalisation of ATP7A and ATP7B. However, some studies have attributed a particularly important role to MBSs 5 and 6 for copper-induced relocalisation, suggesting that these specific MBSs serve a different function from the others.131 242 246 In addition to copper binding, phosphorylation status plays an important role in the regulation of trafficking of ATP7A and ATP7B. Mutation of the aspartic acid residue in the DKTG motif, which prohibits the formation of an acylphosphate intermediate, abolishes copper-induced trafficking of ATP7A and ATP7B from the TGN to the cell periphery.114 140 Conversely, hyperphosphorylation of ATP7A or ATP7B induced by mutation of the TGE motif results in a constitutive peripheral localisation.114 A combination of mutations of the TGE motif, the six amino terminal MBSs and the CPC motif in ATP7B also shows constitutive peripheral localisation.247 This fascinating observation can be explained in two ways. It is possible that other metal-binding sites are present in ATP7B through which relocalisation can be induced by copper. Alternatively, ATP7B constitutively cycles between the TGN and the cell periphery, and copper binding serves as a retention signal, inhibiting retrograde trafficking to the TGN. Retrograde trafficking of ATP7A from the plasma membrane back to the TGN requires a dileucine motif within the carboxyterminal tail.134 248 249 Although this motif might serve as a classic clathrin-mediated endocytosis targeting motif, other mechanisms play a role in the internalisation of ATP7A from the plasma membrane.250 251 A trileucine motif is present at the same position of ATP7B, and although ATP7B does not reach the plasma membrane, mutation of this motif also results in a constitutive peripheral localisation of ATP7B.140
These studies have provided valuable insights into the mechanisms behind copper-induced trafficking of ATP7A and ATP7B. One of the most important remaining questions is how exactly copper is exported from the cell after this event has occurred, and this should be the focus of future studies.
Box 4 The ATP7A and ATP7B interactome
Identification of protein–protein interactions is a powerful tool to understand protein function and how perturbation of protein function results in human disease. Over the past few years several interacting partners for ATP7A and ATP7B have been reported, resulting in a rapid elaboration of the ATP7A and ATP7B interactome (figure 6).177 252–255 The best characterised interacting partner of ATP7A and ATP7B is ATOX1. Through its role to deliver copper to ATP7A and ATP7B, ATOX1 plays an essential role in the function of ATP7A and ATP7B, which is further discussed in the text. However, additional proteins might be involved in the delivery of copper to ATP7A and ATP7B. Among these is glutaredoxin, which interacts with both ATP7A and ATP7B in a copper-dependent manner, and has been hypothesised to regulate copper binding by ATP7A and ATP7B.252 In addition, copper delivery to ATP7A and ATP7B might be regulated by the immunophillin FKBP52. This protein interacts with ATOX1 in a copper-dependent manner, and overexpression of FKBP52 results in increased cellular copper efflux.256 The exact mechanisms through which FKBP52 attenuates its role in the copper-excretion pathway need to be further characterised. COMMD1, the protein defective in CT, interacts with both ATP7A and ATP7B149 177 (de Bie et al, unpublished observations). The function of these interactions remains to be characterised, but the phenotype resulting from absence of COMMD1 in CT suggests that COMMD1 and ATP7B cooperate to facilitate billiary copper excretion (further discussed in the text). Another recently identified interacting partner for ATP7A is AIPP1.255 This PDZ domain-containing protein binds to the carboxy terminal tail of ATP7A, where a putative PDZ binding motif is present. As this motif is required for targeting of ATP7A to the basolateral membrane, it has been suggested that AIPP1 has a regulatory role in the copper-induced trafficking of ATP7A.136 255 Through its amino terminal tail, ATP7B interacts with the dynactin subunit p62 in a copper-dependent manner.253 As the dynactin protein complex is involved in membrane vesicle movement along microtubules, this interaction suggests that the p62 dynactin subunit facilitates copper-induced trafficking of ATP7B.257 Further identification and characterisation of novel interacting partners of ATP7A and ATP7B will also shed new light on previously unanticipated functions of these proteins, as evidenced by the recently observed interaction between ATP7B and a promyelocytic leukemia zinc finger protein (PLZF) isoform. Characterisation of the interaction between the PLZF isoform and ATP7B suggested that ATP7B attenuates activation of the ERK signaling pathway.254 Further investigation is needed to determine the implications of this observation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank members of the Leo Klomp and Cisca Wijmenga laboratories for helpful discussions. The work in these two laboratories is funded by the Netherlands Organization for Scientific Research (Zon-MW, grant 40-00812-98-03106), the Dutch Digestive Diseases Foundation (MLDS, grant WS 02-34), the Wilhelmina Children’s Hospital (WKZ) Fund (grant 901-04-219). The authors declare they have no competing interests.
REFERENCES
Footnotes
Competing interests: None declared.
- Abbreviations:
- A-domain
- actuator domain
- AIPP1
- ATPase interacting PDZ protein 1
- CCS
- copper chaperone for superoxide dismutase 1
- CT
- copper toxicosis in Bedlington terriers
- CTR1
- copper transporter 1
- ER
- endoplasmic reticulum
- ETIC
- endemic Tyrolean infantile cirrhosis
- ICC
- Indian childhood cirrhosis
- ICT
- idiopathic copper toxicosis
- LEC
- Long–Evans cinnamon
- MBS
- metal-binding site
- MD
- Menkes disease
- N-domain
- nucleotide-binding domain
- NF
- nuclear factor
- OHS
- occipital horn syndrome
- OMIM
- Online Mendelian Inheritance in Man
- P-domain
- phosphorylation domain
- PLZF
- promyelocytic leukemia zinc finger protein
- SOD1
- superoxide dismutase 1
- TGN
- trans-Golgi network
- WD
- Wilson disease