Article Text

Abstract
Oesophageal atresia and/or tracheo-oesophageal fistula are relatively common malformations occurring in approximately 1 in 3500 births. In around half of the cases (syndromic oesophageal atresia), there are associated anomalies, with cardiac malformations being the most common. In the remainder (non-syndromic cases), oesophageal atresia/tracheo-oesophageal fistula occur in isolation. Data from twin and family studies suggest that genetic factors do not play a major role, and yet there are well-defined instances of this malformation where genetic factors clearly are important. This is highlighted by the recent identification of no fewer than three separate genes with a role in the aetiology of oesophageal atresia: those for Feingold syndrome (N-MYC), anophthalmia-oesophageal-genital (AEG) syndrome (SOX2), and CHARGE syndrome (CHD7). Additional support for genetic factors in this malformation comes from chromosomal studies and mouse models. This paper reviews current knowledge of the genetics and epidemiology of the different oesophageal atresia/tracheo-oesophageal fistula syndromes and associations.
- anophthalmia-oesophageal-genital
- CHARGE syndrome
- Feingold syndrome
- oesophageal (esophageal) atresia
- tracheo-oesophageal (-esophageal) fistula
Statistics from Altmetric.com
- anophthalmia-oesophageal-genital
- CHARGE syndrome
- Feingold syndrome
- oesophageal (esophageal) atresia
- tracheo-oesophageal (-esophageal) fistula
Oesophageal atresia and tracheo-oesophageal fistula (OA/TOF) are common life-threatening malformations with an incidence of approximately 1 in 3500 births. The aetiology of OA/TOF is unknown in the majority of cases. In approximately half of the cases (syndromic oesophageal atresia), there are other associated anomalies, with cardiac malformations being the most common. These may occur as part of VATER or the VACTERL association (OMIM 192350). In the remaining cases, OA/TOF occur in isolation (non-syndromic oesophageal atresia).
The birth of a child with OA/TOF into a family without a previous history of the condition is associated with a low recurrence risk, of the order of 1%.1 The twin concordance rate for OA/TOF is likewise low, at around 2.5%.2 These data do not indicate a major role for genetic factors in the pathogenesis of OA/TOF, yet there are well-defined instances of the condition where genetic factors are clearly important. Trisomies of chromosomes 18 and 21 are a significant risk factor for OA/TOF, and there are other examples of specific chromosomal imbalances, discussed below, which also predispose to this malformation. Until recently, no genes had been associated with oesophageal atresia in humans. However, three separate genes associated with syndromic OA/TOF in humans have now been identified,3–5 making this an exciting time for those interested in the aetiology of this malformation.
This paper reviews current knowledge of the genetics, aetiology, and epidemiology of syndromic and non-syndromic OA/TOF, placing this new information in its overall context.
EMBRYOLOGY
The intimate relationship of the trachea and oesophagus is evident from early embryonic life. The respiratory diverticulum appears at around 4 weeks of age as an outgrowth from the ventral wall of the foregut and invades the surrounding splanchnic mesoderm.
Although there are many references in the literature6–8 to the separation of the trachea from the foregut by means of a process of “pinching off” achieved by the existence of lateral tracheo-oesophageal ridges which grow and fuse in the midline, Williams et al9 have drawn attention to the lack of any direct imaging or reconstruction through histological sections of the tracheo-oesophageal ridges, and in addition have noted the presence of apoptotic bodies at the point of tracheo-oesophageal separation (originally described by Qi and Beasley10). Both groups of authors suggest that it is the process of apoptosis, with “collapse and fusion of the lateral walls of the foregut”, rather than the growth and fusion of lateral ridges, which causes oesophagus and trachea to separate, although the existence of a tracheo-oesophageal septum arising from fusion of longitudinal ridges within the lumen of the foregut is still postulated by some authors.7
ANATOMIC VARIANTS OF OA/TOF
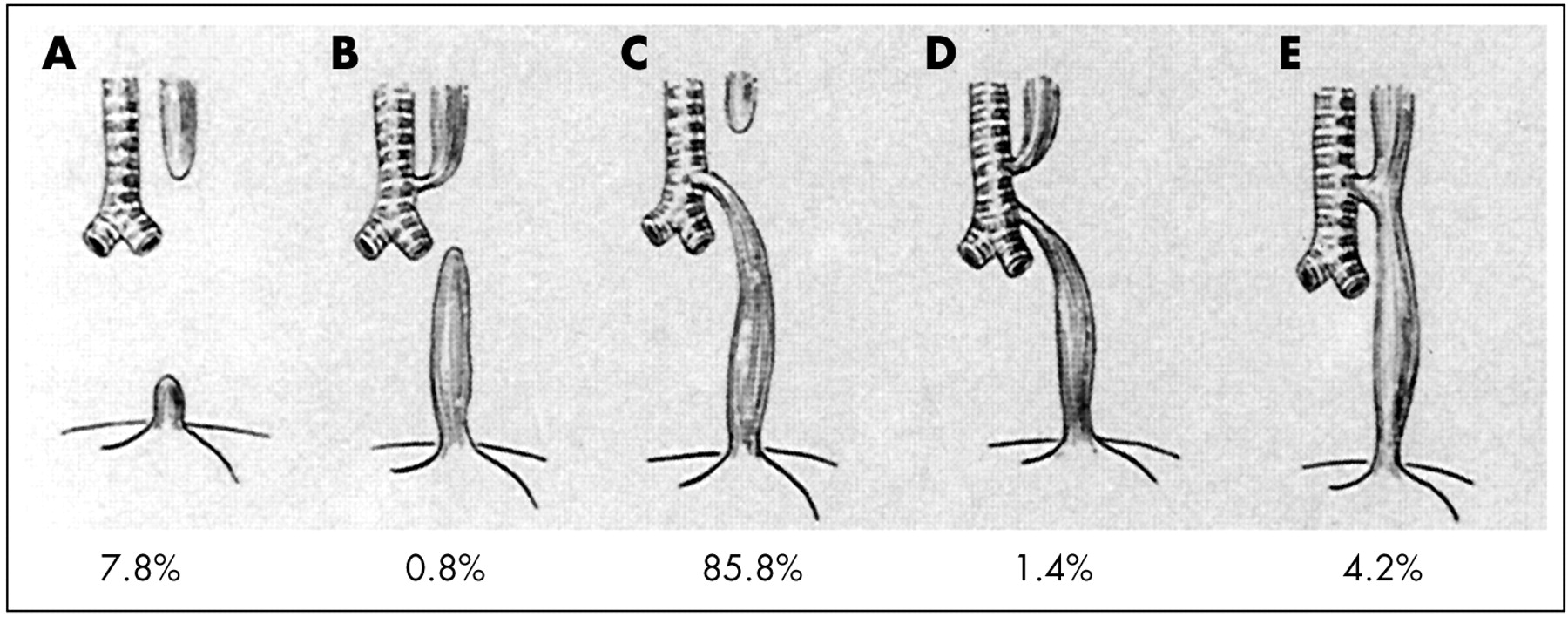
Several anatomic classifications have been proposed (reviewed in Harmon and Coran11); that illustrated in fig 1 is taken from Gross.12 The incidence of the various types of anomaly is derived from more than 2200 cases in six large series.11 A very detailed classification which corresponds broadly to other schemes but includes other rarer anatomic variants such as membranous atresia, oesophago-bronchial fistula, and oesophageal duplication, has been described.13

Anatomical classification of anomalies of the oesophagus and trachea, with approximate incidences. (From: Gross RE. Surgery of infancy and childhood. Philadelphia, PA: WB Saunders, 1953. Reprinted with permission from Elsevier Press.)
EPIDEMIOLOGY
Data from three large scale epidemiological studies are summarised in table 1. The incidence of OA/TOF is approximately 1 in 3500 births. There is little evidence to support significant geographical or secular variation in the incidence of OA/TOF. Robert et al2 reported a significantly lower birth frequency (1.82/10 000 births) in Norway, but the birth frequency in Sweden in the same study was 2.67/10 000, and other published data investigating this trend are lacking. No consistent secular trend was identified in the above studies, although Depaepe et al14 identified a downward trend in birth frequency over time in different regions of Europe. When chromosomal cases are excluded, there is no evidence for a link between OA/TOF and maternal age.
OA/TOF epidemiological data
The figures for isolated versus associated OA/TOF (presence of an additional congenital malformation) vary between 38.7%,15 45.0%,14 and 57.3%2 (chromosomal cases included). Differences in ascertainment are likely to account for some of the discrepancy. For example, in the Worldwide study,2 only those births occurring after 28 weeks’ gestation were included. Many early fetal deaths due to multiple malformations are therefore likely to have been excluded, with the likely effect of inflating the figure for isolated cases.
Data on malformations associated with OA/TOF in these epidemiological studies are summarised in table 1. The malformations most commonly found with OA/TOF are those present in the VACTERL association. These are discussed further in the section on the VACTERL association below.
The data suggest that the incidence of trisomies and other chromosomal imbalances is fairly consistent at between 6 and 10%. The lower figure of 6.3% in the Worldwide study2 is again likely to be explicable on the basis of cases excluded prior to 28 weeks’ gestation. The total number of cases in infants with trisomy 18 (127) exceeds the number of cases due to trisomy 21 (102) despite the fact that trisomy 18 is much rarer than trisomy 21. This appears to indicate that trisomy 18 may be a greater risk factor for OA/TOF than trisomy 21.
OA/TOF AND TWINNING
Congenital malformations, including OA/TOF, occur more commonly in twins. Mastroiacovo et al16 found that, of 92 malformations studied, 39, including OA/TOF, were more common in twins than in singletons. The relative risk for OA/TOF in twins compared with singletons was 2.56 (95% confidence interval 2.01 to 3.25). In one series,17 the frequency of twinning in patients with OA/TOF was 7% compared with the expected population rate of 2.3%.
The concordance rate in twins is low, suggesting that mechanisms other than genetics are responsible for the occurrence of OA/TOF in twins. In the series reported by Robert et al,2 80 twin pairs were identified, of which just two pairs (2.5%) were concordant, the remaining 78 being discordant. Of the 80 pairs, the sex of the co-twin was known in 50. Forty three were like-sexed (22 male, 21 female) and only seven unlike-sexed. These data may suggest an increased incidence of monozygosity in these twin pairs, although confirmatory data are lacking. Other studies have found similar results for twin pairs.18
FAMILY STUDIES OF OA/TOF
Warren et al19 provided the first estimate of offspring risk in patients with OA/TOF. They studied 79 patients who had undergone operations for OA/TOF between 1947 and 1959. Fifteen of these patients had produced a total of 28 children. One of these children had OA/TOF. The sibling recurrence risk was similarly low, with one instance of recurrence in the 130 siblings of the 79 study patients.
McMullen et al20 studied first degree relatives of 140 index cases with OA/TOF, and obtained offspring and sibling recurrence risk figures for VACTERL-type malformations of 2–3% and 1.4%, respectively.
Brown et al21 compared the incidence of VACTERL-type malformations in the first degree relatives of OA/TOF cases versus controls. They found that 5.8% of case families versus 3.1% of control families contained at least one first degree relative with one or more of the extended VACTERL components (p<0.01).
HUMAN SINGLE GENE DISORDERS AND OESOPHAGEAL ATRESIA
Feingold syndrome
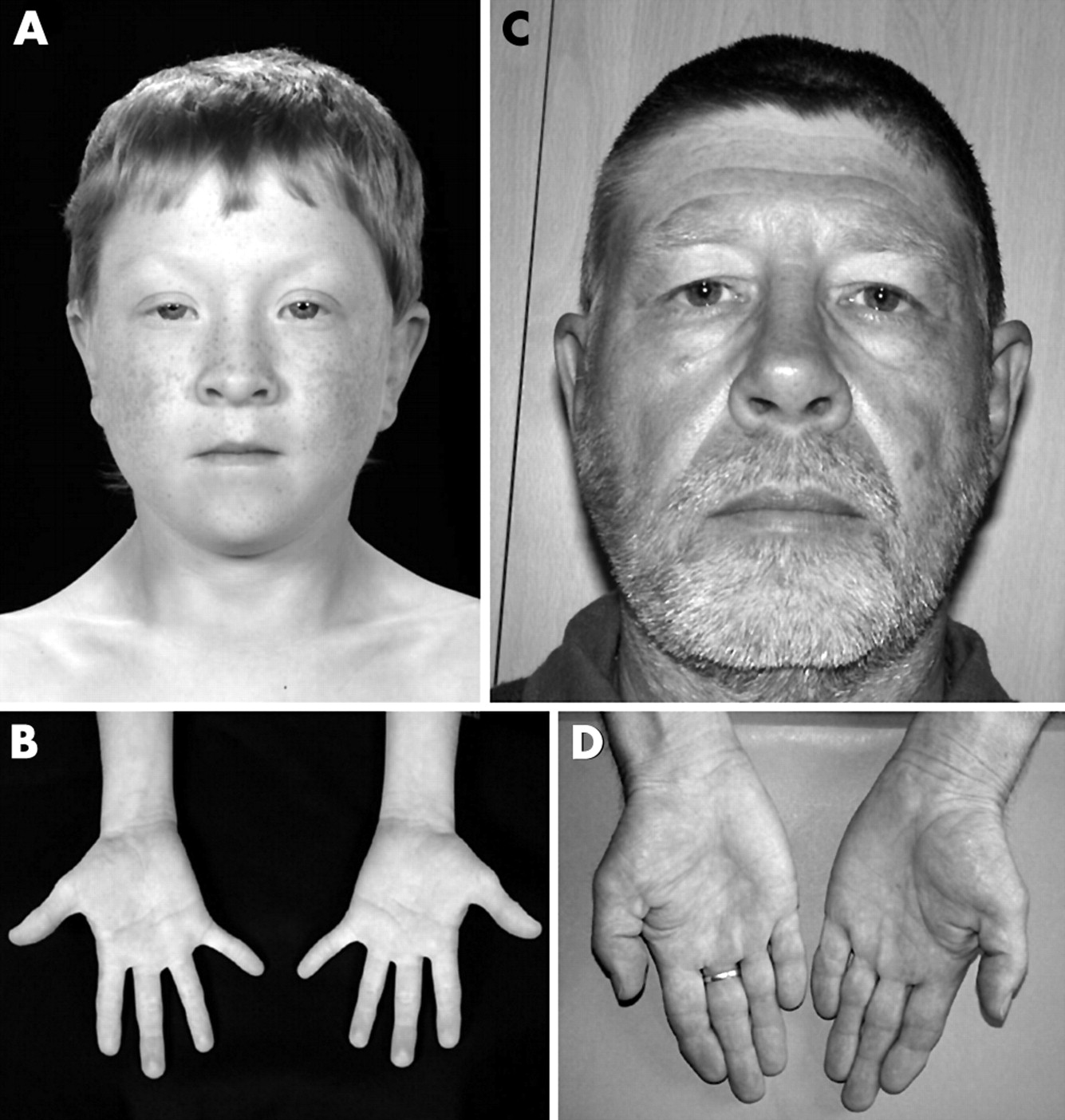
Feingold syndrome (oculodigitoesophagoduodenal syndrome, OMIM 164280; reviewed by Celli et al22) is characterised by microcephaly, digital abnormalities, and atresias of the alimentary tract, principally of the oesophagus. Microcephaly occurs in the majority of cases, and may be associated with usually mild learning disability. The digital anomalies are characteristic, involving brachymesophalangy and clinodactyly of the second and fifth digits in the hands, and variable syndactyly of the toes. An affected father and son are shown in fig 2. Inheritance is autosomal dominant, and the gene for the condition was previously mapped to chromosome 2p23.23

{kind=link}
{kind=link}
Feingold syndrome. Affected boy (A, B) and his father (C, D). Note short palpebral fissures and periorbital fullness (A), mild bilateral fifth finger clinodactyly (B), short palpebral fissures (C), and bilateral clinodactyly of second and fifth fingers with brachymesophalangy of the second fingers (D). (Reproduced with permission from Shaw-Smith C, Willatt L, Thalange N. Clin Dysmorphol 2005;14(3):155–8.)
Recently, mutations in the gene N-MYC were shown to cause Feingold syndrome.3 Amplifications of this gene had previously been associated with neuroblastoma,24 and mice with loss of function mutations in the gene had defects in embryonic organogenesis, although not specifically of the oesophagus or trachea.25,26 N-MYC was not therefore an obvious candidate. Nonetheless, van Bokhoven and co-workers identified 12 different heterozygous mutations and two whole-gene deletions in N-MYC. All of the mutations occurred in exon 3 of the gene. A role for N-MYC as a regulator of progenitor cell proliferation and differentiation in the mouse lung has recently been identified,27 but the role of this gene in foregut and trachea development has yet to be elucidated.
CHARGE syndrome
OA/TOF is not a major criterion for CHARGE syndrome, which comprises Coloboma, Heart defect, Atresia choanae, Retarded growth, Genital hypoplasia and Ear anomalies, but it occurs in about 10% of cases of the syndrome.28 Heterozygous mutations (10) or whole gene deletions (two) of the chromodomain helicase DNA-binding family member CHD7 were recently identified in 12/19 individuals with CHARGE syndrome.4 In a more recent clinical survey, oesophageal atresia and/or tracheo-oesophageal fistula were found in 17% of individuals with CHD7 mutations.29
Chromodomain proteins have a role in the epigenetic regulation of heterochromatin function and euchromatic gene expression. One hypothesised role is that they serve to protect the chromatin fibre from changing its epigenetic state.30 Genotype-phenotype and functional studies in humans and model organisms will help to clarify the relationship between CHD7 and CHARGE syndrome.
Anophthalmia-oesophageal-genital syndrome (AEG syndrome)
The association of oesophageal atresia with anophthalmia and genital abnormalities is a rare though well-documented entity in the literature, with fewer than 20 cases reported.31,32 Loss of function mutations in SOX2 were previously reported in 4/102 patients with microphthalmia, anophthalmia, or coloboma.33 Recently, heterozygous SOX2 mutations have been demonstrated in patients with AEG syndrome.5 Little is known of the role of SOX2 in foregut and lung development. The chick homologue of SOX2, cSox2, is expressed in the developing foregut, though not more caudally in the gut tube. During morphogenesis, cSox2 expression levels fall in the developing lung primordium as it invades the surrounding mesenchyme.34 The precise role of SOX2 in relation to the pathogenesis of OA/TOF remains to be elucidated.
Fanconi anaemia and OA/TOF
OA/TOF, as well as other gastro-intestinal atresias (anal, duodenal) is a well-documented component of the malformation spectrum associated with Fanconi anaemia. Gastro-intestinal atresias have been reported in 14% of cases of Fanconi anaemia.35 Other malformations falling within the VACTERL spectrum also occur: skeletal (71%), cardiac (13%), and renal (34%).35 The birth frequency of OA/TOF is approximately 1 in 3000, that of Fanconi anaemia approximately 1 in 300 000, and that of Fanconi anaemia presenting with a gastro-intestinal atresia another order of magnitude lower. Clearly, therefore, the prior risk of FA in a child born with OA/TOF must be very small, and the contribution of FA to the aetiology of OA/TOF likewise small. Nonetheless, it is possible that mutations in one of the FANC genes are responsible for OA/TOF in a proportion of individuals with this malformation. Some of these individuals may not be penetrant for the haematological phenotype. Now that the majority of the FANC genes have been identified,36,37 this hypothesis is testable. The possible aetiological basis for the relationships between DNA repair disorders and human malformations has recently been discussed.38
Multiple gastro-intestinal atresias (MGIA)
There are several reports of a lethal syndrome of multiple gastro-intestinal atresias in the oesophagus, small intestine, and biliary system.39–41 Parental consanguinity and sibling recurrence in these families point to autosomal recessive inheritance. No mapping studies have been performed to date. This condition appears to be distinct from the syndrome of hereditary multiple intestinal atresias (HMIA), also considered to be autosomal recessive,42 which does not involve the oesophagus. In HMIA, septal atresias with incomplete separation of the atretic segments occur throughout the stomach and large bowel.
Oesophageal atresia and chromosomal abnormalities
The association between OA/TOF and trisomy for chromosomes 18 and 21 has already been discussed above in the section on epidemiology.
There is a weak association between OA/TOF and the 22q11 deletion syndrome. Digilio et al43 found a single case of 22q11 deletion syndrome in a series of 15 patients with syndromic oesophageal atresia, and there are other case reports in the literature.44
There appears to be a link between OA/TOF and deletions at chromosome 17q22q23.3. Marsh et al45 reported a single case and reviewed two others, in all three of whom OA/TOF and congenital heart defects were present. In addition, these patients had dysmorphic facial features and minor skeletal malformations. This chromosomal region contains the genes TBX2 and TBX4. Mice haploinsufficient for the Tbx2 gene have been shown to have atrioventricular septal defects, though not OA/TOF.46 A study in chick47 showed that ectopic expression of Tbx4 in foregut visceral mesoderm caused failure of formation of tracheo-oesophageal septum. Hitherto, haploinsufficiency of or mutations in these two genes have not been associated with OA/TOF in humans, except in the context of the large chromosomal deletions discussed above.
Patients with deletions at chromosome 13q32 have been reported to have multiple elements of the VACTERL association. This possible link is discussed further in the section below on the VACTERL association.
Disruption of the gene BPAG1 was recently reported in an individual with a 6;15 reciprocal translocation and oesophageal atresia.48
OESOPHAGEAL ATRESIA AND TERATOGENS
Clementi et al49 reported a case of congenital anomalies, including oesophageal atresia, in a child exposed to methimazole in the first trimester of pregnancy and drew attention to two previous cases with the same association.50
THE VACTERL ASSOCIATION
OA/TOF is a component of the VACTERL association, which comprises Vertebral defects, Anal atresia, Cardiac defects, Tracheo-Esophageal fistula, Renal malformations, and Limb defects. The association was first proposed by Quan and Smith in 1972,51 initially as the acronym VATER, where the letter “R” stood for “radial dysplasia”. Subsequently, the acronym was expanded to include cardiac and renal defects.52 Both terms remain in use in the literature, illustrating the problem of defining the clinical features that comprise the association. In this review, the acronym VACTERL is used except where VATER is appropriate for historical reasons.
Earlier studies53–57 confirmed the non-random occurrence of the association but did not specifically address the question of which of the more peripheral elements of the association should be included within its definition. More recent studies58 have attempted to address this issue by taking, as far as possible, an approach free of ascertainment bias. The results58 indicated a distinct group of malformations corresponding with the VACTERL association, with an “upper” group associated with heart malformations and a “lower” group associated with renal malformations. The authors acknowledged the difficulty of controlling for possible bias in the way that patients are investigated which might tend to confirm some associations (oesophageal atresia, upper costo-vertebral defects, cardiac defects), but not others where the anatomic locations are more disparate.
There are very few published series of VACTERL patients in which the clinical phenotypes have been carefully delineated, perhaps unsurprisingly in view of the difficulties of definition, and the risk of inadvertent inclusion of cases with a syndromic diagnosis. Weaver et al59 reported a series of 46 patients.
One of the key difficulties in studies of this nature is the evolving nature of our knowledge of syndromes which may overlap with or resemble the VACTERL association. Feingold syndrome, CHARGE syndrome, 22q11 deletion syndrome, Townes-Brocks syndrome, and Pallister-Hall syndrome (all discussed further below) show some phenotypic overlap with the VACTERL association, and most series of patients ascertained by epidemiological means are not divided into subgroups according to confirmed or possible syndromic diagnoses.
Individuals with the VACTERL association do not typically have facial dysmorphic features, learning disability, or abnormalities of growth, including head circumference. For these individuals, sibling and offspring recurrence risks are low, and are usually quoted as being around 1%. There are very few instances of recurrence of the VACTERL association in the literature. However, two examples are Nezarati and McLeod60 and Auchterlonie and White.61
Where dysmorphic features, growth abnormalities, and/or learning disability are present, a syndromic diagnosis or chromosomal abnormality may be the underlying cause, as discussed in the following section.
SINGLE GENE DISORDERS RESEMBLING THE VACTERL ASSOCIATION
An overview of single gene disorders with features in common with the VACTERL association is presented in table 2.
Phenotypic overlap with the VACTERL association: disorders of known genetic aetiology
Feingold syndrome
The overlap between Feingold syndrome and the VACTERL association has been discussed in detail by Celli et al.22 As well as oesophageal atresia which occurs in around 25% of affected individuals, other VACTERL-type malformations occur in Feingold syndrome, including vertebral malformations, anal atresia, and cardiac, renal, and limb malformations.22 There are also significant differences between the two conditions: Feingold syndrome is dominantly inherited, whereas the VACTERL association occurs sporadically. Microcephaly, minor facial dysmorphism, and learning difficulties occur in Feingold syndrome but not classically in the VACTERL association. Van Bokhoven et al3 tested eight individuals with the VACTERL association; no mutations in the N-MYC gene were identified.
CHARGE syndrome
CHARGE syndrome may cause diagnostic difficulty, particularly where few features are present and the more characteristic features of the syndrome (coloboma, choanal atresia) are absent. Identification of the causative gene4 means that it will be possible to delineate the full phenotypic spectrum. It is likely that some cases previously classified clinically as the VACTERL association will prove to have CHD7 mutations, although this remains to be seen.
Fanconi anaemia and the VACTERL association
The apparently Mendelian segregation of VACTERL in association with hydrocephalus (VACTERL-H) is a rarely reported but distinct entity. X linked66–68 and recessive forms69 have been described.
Cox et al69 reported a dizygotic twin pair with features of VACTERL-H and no evidence of increased chromosome breakage on routine testing. Homozygous mutations in the FANCC gene were detected in both twins, providing the first molecular evidence of a link between VACTERL-H and Fanconi anaemia.
OA/TOF, as well as duodenal atresia, have been reported in several family members in pedigrees with VACTERL-H and apparently X linked inheritance. Some of these families have contained individuals with an increased incidence of chromosome breakages, diagnostic of Fanconi syndrome. The family reported by Wang et al66 contained three such individuals; but breakages were not found in a single case in a pedigree containing four affected individuals reported by Lomas et al.68 A gene responsible for X linked Fanconi anaemia has recently been identified;37 it will be interesting to test the possible role of this gene in X linked VACTERL-H families.
In one series, VACTERL-type clinical presentations accounted for 5% of all cases of Fanconi anaemia and there was over-representation of complementation groups D1, E, and F in this group.70 Faivre et al71 reported a series of 13 FA cases presenting with VACTERL-type malformations, all of whom had radial ray anomalies, and 12 out of 13 of whom had an additional FA feature (café au lait patches, microcephaly, growth retardation, or dysmorphism). They suggested that chromosome breakage studies should be performed in patients in this clinical group.
Townes-Brocks syndrome
Townes-Brocks syndrome (reviewed in Powell and Michaelis64) is an autosomal dominant disorder, the main manifestations of which are external ear anomalies, preaxial polydactyly, imperforate anus, and renal malformations. Oesophageal atresia is not a reported feature; there may be an association with congenital heart disease but this is controversial.64 The most common limb defects are triphalangeal thumb and preaxial polydactyly. Hypoplasia of the thumb and radial bone are not features of this syndrome. Structural vertebral anomalies have not been reported. Mutations in the SALL1 gene at 16q12.1 are responsible for Townes-Brocks syndrome.72
Pallister-Hall syndrome
Anal atresia and limb malformations are features of this dominantly inherited syndrome,65 though in practice there should be little difficulty in distinguishing it from the VACTERL association as hypothalamic hamartoma and neurological complications are cardinal features, and the limb malformations are not of the VACTERL type. Pallister-Hall syndrome is due to mutations in the GLI3 gene,65 and, given the mouse knockout phenotypes discussed below, it is of interest that laryngeal clefts and atypical lung lobation are also features of this syndrome.
OTHER CONDITIONS WITH PHENOTYPIC OVERLAP WITH THE VACTERL ASSOCIATION
22q11 deletion syndrome may show significant clinical overlap with the VACTERL association. Oesophageal atresia has been reported43 and may occur in combination with cardiac and renal malformations, though limb and vertebral anomalies and anal atresia are not recognised features.
There is a recognised overlap between the VACTERL association and hemifacial microsomia or oculo-auriculo-vertebral spectrum.73,74 Although ear and eye malformations are typical of hemifacial microsomia, vertebral anomalies, congenital heart defects, and oesophageal atresia are also described.74,75 The aetiology of hemifacial microsomia is poorly understood. Familial cases are described and linkage to chromosome 14q32 has been assigned on the basis of a large dominant pedigree.76
There are three reports in the literature of an association between VACTERL and tibial aplasia.77,78 All three cases had OA/TOF, but additional features (malformed ears, cleft lip/palate) were present in one case.
Reardon et al79 reported a mutation in the PTEN gene in a patient with macrocephaly, tracheo-oesphageal fistula, and bilateral thumb hypoplasia.
TERATOGENS ASSOCIATED WITH THE VACTERL ASSOCIATION
A two- to threefold increase in the incidence of multiple congenital malformations is recognised in the children of diabetic mothers.80 Most characteristically, these malformations appear to affect the caudal part of the body, although central nervous system anomalies, oesophageal atresia, congenital heart defects, and rib and vertebral anomalies are all described, indicating a clear overlap with the VACTERL association.
CHROMOSOMAL IMBALANCES IN THE VACTERL ASSOCIATION
Walsh et al81 reported a case of peno-scrotal transposition associated with all six VACTERL components, in a patient with distal chromosome 13q deletion. Peno-scrotal transposition is a rarely noted finding in the VACTERL association.82 Walsh et al81 performed a detailed review of the literature on deletions of the distal long arm of chromosome 13 and identified 147 patients with this chromosomal abnormality. Of these, five had four VACTERL elements, 12 had three elements, 23 had two elements, and 28 had one element, although only one patient, that reported by Walsh et al,81 had OA/TOF (oesophageal atresia and a tracheo-oesophageal fistula in this instance). As well as components of the VACTERL association, some of these patients also had CNS malformations, including holoprosencephaly, anencephaly, hydranencephaly, callosal agenesis, neural tube defects, and ventriculomegaly; in addition there were often other features such that the diagnosis of the VACTERL association was not postulated in the original publications.83–86 The review included some patients previously investigated in a study by Brown et al87 which had identified a critical region for major malformations at chromosome 13q32. Mutations in the ZIC2 gene, which is at chromosome 13q32, are associated with holoprosencephaly, suggesting that CNS malformations in patients with distal 13q deletions may be due to haploinsufficiency of this gene.
Cinti et al88 reported a supernumerary ring chromosome 12 in a patient with an anorectal malformation, vertebral anomalies, and an absent kidney. There are very few other reports of chromosomal abnormalities in patients with the VACTERL association.89,90
OA/TOF, VACTERL, AND ASSOCIATIONS
The term “association” was first used specifically with respect to the VATER association by Quan and Smith.51 Lubinsky91 gave the following definition of associations: “Associations are derivatives of causally non-specific disruptive events acting on developmental fields”. In the case of errors of blastogenesis, the developmental field constitutes the entire embryo.92 Opitz92 gave a modified definition of associations: “Associations represent the idiopathic occurrence of multiple congenital anomalies during blastogenesis” (italics here and later in this section are those of CS-S). The key concept is the implication that different causal factors acting at particular stages in development give rise to similar patterns of malformations. Martinez-Frias et al93 advocated the use of the term polytopic field defect to replace the term association in this context. In the case of the VACTERL association, causative perturbations may be of known genetic origin, in which case they give rise to syndromes (see table 2), and the term association is no longer applicable; or they may be due to environmental agents (teratogens), in which case they remain idiopathic. The underlying causes of the sporadic forms of the VACTERL association for the most part remain to be elucidated, with the exception of those instances due to maternal diabetes. According to the model proposed by Lubinsky, single malformations derived from teratologic events can be part of the same spectrum as associations.91 In this sense, oesophageal atresia with or without tracheo-oesophageal fistula, and sporadic forms of the VACTERL association can, for the purposes of hypothesis testing, be considered to be two different points on a spectrum constituting a single overarching entity.
KNOCK-OUT MICE FEATURING TRACHEO-OESOPHAGEAL MALFORMATIONS
Mice deficient in genes in the sonic hedgehog pathway, including sonic hedgehog itself, exhibit OA/TOF. Mice homozygously deficient for sonic hedgehog display multiple malformations including oesophageal atresia/stenosis and tracheo-oesophageal fistula.94–96 Mice homozygously deficient for the Gli2 gene likewise exhibit multiple anomalies, including a foregut defect consisting of hypoplastic oesophagus and trachea with oesophageal stenosis.97 Haploinsufficiency of Gli3 on the Gli2−/− background produces a more severe defect with OA/TOF.97 Gli2−/− Gli3−/− mice have a severe phenotype with absence of oesophagus, trachea, and lungs.97
The winged helix transcription factor foxf1 is activated by sonic hedgehog signalling and mice haploinsufficient for foxf1 display narrowing of the oesophagus and trachea, oesophageal atresia, and tracheo-oesophageal fistula.98
There is one report describing oesophageal abnormalities associated with the targeted disruption of a homeobox gene. Mice haploinsufficient for the homeobox gene hoxc4 had complete occlusion of the oesophageal lumen, with extensive disorganisation of the oesophageal musculature, although not oesophageal atresia.99
Mice homozygously deficient for the homeodomain transcription factor nkx2.1 show a severe tracheo-oesophageal phenotype with a common lumen that connects the pharynx to the stomach. In addition, the lungs are profoundly hypoplastic with absence of distal structures.100 Finally, failure of separation of the oesophagus and trachea is seen in mouse embryos doubly homozygous for mutations in the retinoic acid receptor genes RARα and RARβ2.101
MOUSE MODELS OF THE VACTERL ASSOCIATION
Elements suggestive of, if not identical to, those seen in the VACTERL association are also present in some of the mouse models.102 Shh−/− mice display, in addition to the defects of trachea and oesophagus, absence of the vertebral column, persistent cloaca with abnormalities of heart looping, solitary kidney, and truncating limb malformations. Gli2−/− mice have absent vertebral bodies,103 imperforate anus,104 and limb hypoplasia,103 though no renal or cardiac abnormalities. Gli3−/− mice have abnormal development of neural arches,103 anal stenosis,104 and polydactyly,103 though no renal, cardiac, or tracheo-oesophageal malformations. Gli2−/− Gli3+/− mice display lack of a vertebral column, persistent cloaca, heart looping defects, horseshoe kidney, and polydactyly.102
In humans, mutations in SHH are associated with holoprosencephaly,105 and mutations in GLI3 with Pallister-Hall syndrome.65 There are no published data on the possible roles of these genes in patients with syndromic OA/TOF, to the author’s knowledge, although laryngeal clefts, which may be associated with tracheo-oesophageal fistula, are a feature of Pallister-Hall syndrome.65
OTHER ANIMAL MODELS OF OESOPHAGEAL ATRESIA
Administration of adriamycin to rats at embryonal day 10.5 results in oesophageal atresia and other malformations including those of the VACTERL association.106 Abnormalities of the notochord occur in this model,107 and at the molecular level, abnormalities of expression of genes in the sonic hedgehog pathway have been described.108,109
EVALUATION OF THE PATIENT WITH OA/TOF
Oesophageal atresia is, both clinically and aetiologically, a very heterogeneous condition, and this review highlights the diversity of aetiological factors that can lead to this malformation. The following considerations may aid in diagnosis and in genetic counselling:
-
History: exposure to teratogens, particularly methimazole, in pregnancy; enquire specifically about maternal diabetes in pregnancy.
-
Family history with special attention to individuals in the family with OA/TOF, other GI atresia, cardiovascular or renal malformations, late pregnancy losses, early post-natal deaths, polyhydramnios, microcephaly, or learning disability. All of these may suggest Feingold syndrome.
-
Clinical examination with special attention to head circumference and examination of digits (microcephaly and digital malformations as described in fig 1 may indicate Feingold syndrome), examination of eyes (coloboma may suggest CHARGE syndrome; anophthalmia/microphthalmia may point to AEG syndrome). Dysmorphic features and retarded growth may point to CHARGE syndrome or a chromosomal imbalance such as a trisomy or 22q11 deletion syndrome. Features suggestive of Fanconi anaemia (café au lait patches, microcephaly, growth retardation) should specifically be sought.71
-
Investigations: karyotype for the exclusion of trisomies 18 and 21, FISH for 22q11 deletion (especially when a cardiac defect is present), array-CGH where a chromosomal abnormality is suggested by the clinical features and karyotype and FISH studies are normal. Chromosome breakage studies for Fanconi anaemia should be considered where a VACTERL-type presentation is combined with other suggestive features (see above). Mutation analysis of N-MYC, CHD7, or SOX2 may be sought where the phenotype is suggestive. All patients with OA/TOF should have an echocardiogram; limb or vertebral X rays may be performed according to the clinical picture; renal USS is suggested where there are other features of the VACTERL association.
Genetic counselling
The identification of genes associated with oesophageal atresia offers the prospect of improvements in genetic counselling for this disorder. Even after successful surgical treatment, OA/TOF is associated with significant morbidity and where associated with other congenital malformations, there is appreciable mortality.110 Many parents of affected children, and affected individuals who would like to have children, will welcome the prospect of improved genetic diagnosis that the identification of the Feingold syndrome gene in particular is likely to bring.
The combination of OA/TOF with microcephaly and digital malformations makes Feingold syndrome highly recognisable if the diagnosis is considered, but easily missed if these subtle findings are not specifically sought. Other clinical features may occur; a detailed review is provided by Celli et al.22
The figure of approximately 1% for sibling and offspring recurrence risk, which comes from the study of Warren et al,19 is still quoted in the literature1 although McMullen et al20 gave a slightly higher figure of 2–3% for offspring recurrence risk. Neither study makes reference to the possibility of Feingold syndrome as a possible explanation for familial recurrence of oesophageal atresia. In the earlier study,19 sibling and offspring recurrences occurred in the same family, and it is possible that this family represents an example of Feingold syndrome. In any family where there is a recurrence of OA/TOF, Feingold syndrome should be considered.
For the VACTERL association, the quoted recurrence risk appears low.1 One study20 found an increased incidence of VACTERL-type malformations in first degree relatives of individuals with OA/TOF versus controls, but the recurrence risk specifically for those individuals with the VACTERL association has not been formally studied to the author’s knowledge.
DISCUSSION
The recent identification of three separate genes with a role in the genesis of OA/TOF is exciting for those interested in understanding the aetiology of this malformation, and for those who provide genetic counselling. Available epidemiological and family data do not point to a strong role for genetic factors in OA/TOF. The recurrence risk is quoted at 1% or less,1 and the twin concordance rate is very low, of the order of 2.5%.2 These data appear to suggest that, in the majority of cases, the aetiological factors in this malformation are non-genetic. Against this, the existence of familial forms of oesophageal atresia have been known for many years, and likewise ample data, reviewed above, indicate that chromosomal imbalances may predispose to its occurrence. In mice, evidence for the involvement of genes in the sonic hedgehog pathway in normal oesophageo-tracheal development is very strong, and it will be interesting to study the possible contribution of genes in this pathway to malformations of these structures in humans.
It is clear that these new data present an opportunity for further teasing out the aetiological mechanisms in OA/TOF. As van Bokhoven et al3 suggest, the role of N-MYC in other forms of OA/TOF besides Feingold syndrome should be explored. The same considerations apply to the CHD7 and SOX2 genes, and, further, a “panel” of oesophageal atresia genes can be envisaged which could also include the Fanconi genes as well as members of the sonic hedgehog pathway, for the high-throughput screening of patients with syndromic forms of oesophageal atresia. As well as looking for mutations in the coding sequences of these genes, it will be important to search for copy number changes. The new technique of exon based array CGH111 provides an opportunity to examine copy number changes in a large number of exons simultaneously, thus making high-throughput screening an attractive possibility.
It is likely that the array based technologies which have been successful in identifying submicroscopic chromosomal imbalances in individuals with learning disability and dysmorphic features112,113 will add to the figure of approximately 10% of OA/TOF due to chromosomal imbalances. Two approaches will be useful here: the refinement of existing loci by use of high resolution arrays; and the discovery of new imbalances by means of whole genome array CGH at 1 Mb or tile path resolution. Children with OA/TOF due to a chromosomal imbalance often have a severe phenotype, with significant mortality particularly when other malformations and low birth weight are present.110 This includes children with trisomies 13 and 18 as well as other chromosomal imbalances reviewed above. As well as checking the G-banded karyotype in these children, it will be increasingly helpful to store a DNA sample for future genetic studies.
Concerning embryological studies, there is further scope for detailed molecular and cellular studies of the process of separation of trachea and oesophagus in model organisms. As yet there have been no concerted studies in model organisms to try to characterise the transcriptional regulatory pathways involved in this process, as has been carried out for the early stages of liver development, for example.114,115 Studies of this nature are now feasible and are likely to identify other genes that play a role in this important stage of development.
The refinement of our knowledge of genetic aetiologies in OA/TOF is likely to have implications for future epidemiological studies. One goal of epidemiological research in this area is to try to identify geographical or temporal trends in the birth frequency of OA/TOF in order to gain clues about possible contributing environmental factors. It is likely that the data in previous studies have been confounded by inclusion of individuals with unappreciated genetic aetiologies. These patients could be excluded from future studies of the epidemiology of oesophageal atresia, helping to refine our currently very limited understanding of the non-genetic factors which may contribute to this malformation.
Most significantly, the identification of new genes with a role in the pathogenesis of oesophageal atresia will help to stimulate further research in, and provide genetic counselling for, this distressing and still poorly understood malformation.
Acknowledgments
Thanks to Vicki Martin for comments on the manuscript. Many thanks to Joan Green and Andrew King for library assistance.
The author is grateful to Tracheo-Oesophageal Fistula Support (TOFS UK), the Addenbrooke’s Hospital Charities Committee, and the Wellcome Trust for financial support.
REFERENCES
Footnotes
-
Published Online First 21 November 2005
-
Competing interests: none declared