Article Text

Abstract
Background: A novel autosomal recessive condition, dilated cardiomyopathy with ataxia (DCMA) syndrome, has been identified in the Canadian Dariusleut Hutterite population, characterised by early onset dilated cardiomyopathy with conduction defects, non-progressive cerebellar ataxia, testicular dysgenesis, growth failure, and 3-methylglutaconic aciduria.
Objective: To map DCMA syndrome and identify the mutation underlying this condition.
Methods: A genome wide scan was undertaken on consanguineous Hutterite families using a homozygosity mapping approach in order to identify the DCMA associated chromosomal region. Mutation analysis was carried out on positional candidate genes in this region by sequencing. Reverse transcriptase polymerase chain reaction and bioinformatics analyses were then used to characterise the mutation and determine its effect on the protein product.
Results: The association of DCMA syndrome with a 2.2 Mb region of chromosome 3q26.33 was found. A disease associated mutation was identified: IVS3-1 G→C in the DNAJC19 gene, encoding a DNAJ domain containing protein of previously unknown function (Entrez Gene ID 131118).
Conclusions: The DNAJC19 protein was previously localised to the mitochondria in cardiac myocytes, and shares sequence and organisational similarity with proteins from several species including two yeast mitochondrial inner membrane proteins, Mdj2p and Tim14. Tim14 is a component of the yeast inner mitochondrial membrane presequence translocase, suggesting that the unique phenotype of DCMA may be the result of defective mitochondrial protein import. It is only the second human disorder caused by defects in this pathway that has been identified.
- DCM, dilated cardiomyopathy
- DCMA, dilated cardiomyopathy with ataxia
- LQTS, long QT syndrome
- 3-MGA, 3-methylglutaric acid
- 3-MGC, 3-methylglutaconic acid
- mitochondrial protein import
- dilated cardiomyopathy
- 3-methylglutaconic aciduria
Statistics from Altmetric.com
- DCM, dilated cardiomyopathy
- DCMA, dilated cardiomyopathy with ataxia
- LQTS, long QT syndrome
- 3-MGA, 3-methylglutaric acid
- 3-MGC, 3-methylglutaconic acid
Dilated cardiomyopathy with ataxia (DCMA) syndrome is a novel autosomal recessive disorder found in the Dariusleut Hutterite population, an endogamous population of the great plains region of Canada and the northern United States.1 The major clinical features of DCMA include a severe, early onset dilated cardiomyopathy (DCM), sometimes accompanied by long QT syndrome (LQTS). Prenatal or postnatal growth failure is universally seen, as is a cerebellar syndrome with ataxia, causing significant motor delays. Male genital anomalies are also frequently seen, and range from isolated cryptorchidism to severe perineal hypospadias. In addition, many patients have significant increases in urine organic acids, particularly of 3-methylglutaconic acid (3-MGC) and 3-methylglutaric acid (3-MGA). Additional features include optic atrophy, a mild increase in hepatic enzymes with microvesicular hepatic steatosis, a normochromic microcytic anaemia, and mild to borderline non-progressive mental retardation. The combination of clinical features varies among patients; however, cardiac involvement including DCM and LQTS, cerebellar ataxia, and raised levels of 3-MGC and 3-MGA are the hallmarks of DCMA syndrome.
Initially, DCMA syndrome presents in a very similar manner to Barth syndrome (type II 3-methylglutaconic aciduria), which is an X linked disorder caused by mutations in the TAZ gene and consists of DCM, raised 3-MGC and 3-MGA levels, skeletal myopathy, neutropenia, and growth failure.2 The lack of skeletal myopathy and immunological features, however, as well as the cerebellar features and the autosomal recessive inheritance, clearly distinguishes DCMA from Barth syndrome. DCMA syndrome also shares some resemblance to the other two defined classes of 3-methylglutaconic acidurias; however, the levels of 3-MGC and 3-MGA are too low in DCMA to suggest a primary block in the 3-methylglutaconyl-CoA hydratase enzyme as in type I 3-methylglutaconic aciduria, which is the result of mutations in the AUH gene.3 Cerebellar ataxia is a prominent feature in Costeff optic atrophy syndrome (type III 3-methylglutaconic aciduria), a disorder of the Iraqi Jewish population resulting from mutations in the OPA3 gene. As the name suggests, early onset bilateral optic atrophy is the most prominent feature of this disorder and none of the cardiac features of DCMA syndrome has been noted in this disorder.4 Optic atrophy has been seen in a few patients with DCMA syndrome; however, it is not considered to be one of the major features of DCMA. Type IV 3-methylglutaconic aciduria includes all other cases of 3-methylglutaconic aciduria that cannot be included in types I, II, or III. Neurological, motor, and sensory defects are common within this category, as well as cases of DCM and liver dysfunction. Cases included within the category probably result from various different causes and defects, as increases in both 3-MCG and 3-MGA have been reported in several inborn errors of metabolism and also during pregnancy.5 All cases of DCMA that have been identified thus far are from the Dariusleut Hutterite population; however, it is possible that other cases have been included within this category for lack of a better classification, and thus DCMA syndrome represents an additional category of the 3-methylglutaconic acidurias.
The characteristics of endogamous populations make it possible to map genetic conditions using a homozygosity mapping approach.6 By this approach, we aimed to map DCMA syndrome by identifying regions of the genome that are identical by descent between affected patients from different consanguineous families. Here we report the association of DCMA syndrome with a 2.2 Mb segment of 3q26.33 and the identification of a splice mutation in a novel gene DNAJC19 in DCMA patients. The DNAJC19 protein shares similarity with the yeast TIM14 protein, a component of the mitochondrial protein import system, and DCMA syndrome may represent only the second human disorder associated with defects in mitochondrial protein import.
METHODS
Patients and controls
This study was approved by the conjoint health research ethics board at the University of Calgary. Informed consent was obtained from patients and family members, and genomic DNA was extracted from either cultured fibroblasts or peripheral blood leucocytes by standard phenol chloroform extraction. Fibroblast cells were cultured in MEM medium (Gibco, Gaithersburg, Maryland, USA), with 10–12% fetal bovine serum at 37°C and 5% CO2. A set of 250 genomic DNA samples of various ethnicities was used to check whether the DNAJC19 IVS3-1 G→C mutation exists as a polymorphism in the general population. Genomic DNA from these controls was extracted from peripheral blood lymphocytes by a standard phenol chloroform procedure.
Genome scan
Using a homozygosity mapping approach,6 a complete genome scan was undertaken by genotyping 400 microsatellite markers covering the entire genome to an average density of 10 cM using the ABI PRISM MD-10 linkage mapping set, version 2 (Applied Biosystems, Foster City, California, USA). The genome scan was carried out on five severely affected Hutterite patients meeting inclusion criteria defined by at least two of the following features: dilated cardiomyopathy, 3-methylglutaconic aciduria, and genital anomalies. Fine mapping around potential candidate regions was done by selecting additional microsatellite markers from the ABI PRISM HD-5 Linkage Mapping Set (Applied Biosystems), and NCBI STS map, and by genotyping additional patients and unaffected family members. UniSTS accession numbers for these markers are provided in the electronic database information section below.
Mutation screen
Mutation analysis of candidate genes was undertaken by bidirectional sequencing of predicted exons and splice junctions from genomic DNA from an affected patient, an obligate carrier, and a non-carrier, as determined by the pattern of inheritance of the disease associated haplotype. Sequence data were analysed by subtraction analysis using the Staden package. Entrez Gene entries for positional candidate genes are provided in the electronic database information section.
The DNAJC19 gene was polymerase chain reaction (PCR) amplified in three segments using the following primers (letters in brackets indicate primer positions shown in fig 1B): segment 1, corresponding to exon 6 (1263 base pairs (bp)), forward (P): 5′-GAAGTTTAGACGGTAGGTAGTATAA, reverse (I): 5′-GCTAAATCTCCCTCAGATAAG; segment 2, corresponding to exons 4 and 5 (1298 bp), forward (H): 5′-ATTATCCCATTAATAACTATTGGTC, reverse (E): 5′-CTAAAATACCTCGGGAATTG; and segment 3, corresponding to exons 1, 2 and 3 (1955 bp), forward (D): 5′-CAGGAGAATGGGTCCAAAGCAATCA, reverse (A): 5′-CCGCCCACCCTTCAACCAAC. Amplifications were carried out using the Platinum Taq buffer system (Invitrogen, San Diego, California, USA) with 2.5 mM magnesium chloride, and an annealing temperature of 60°C.

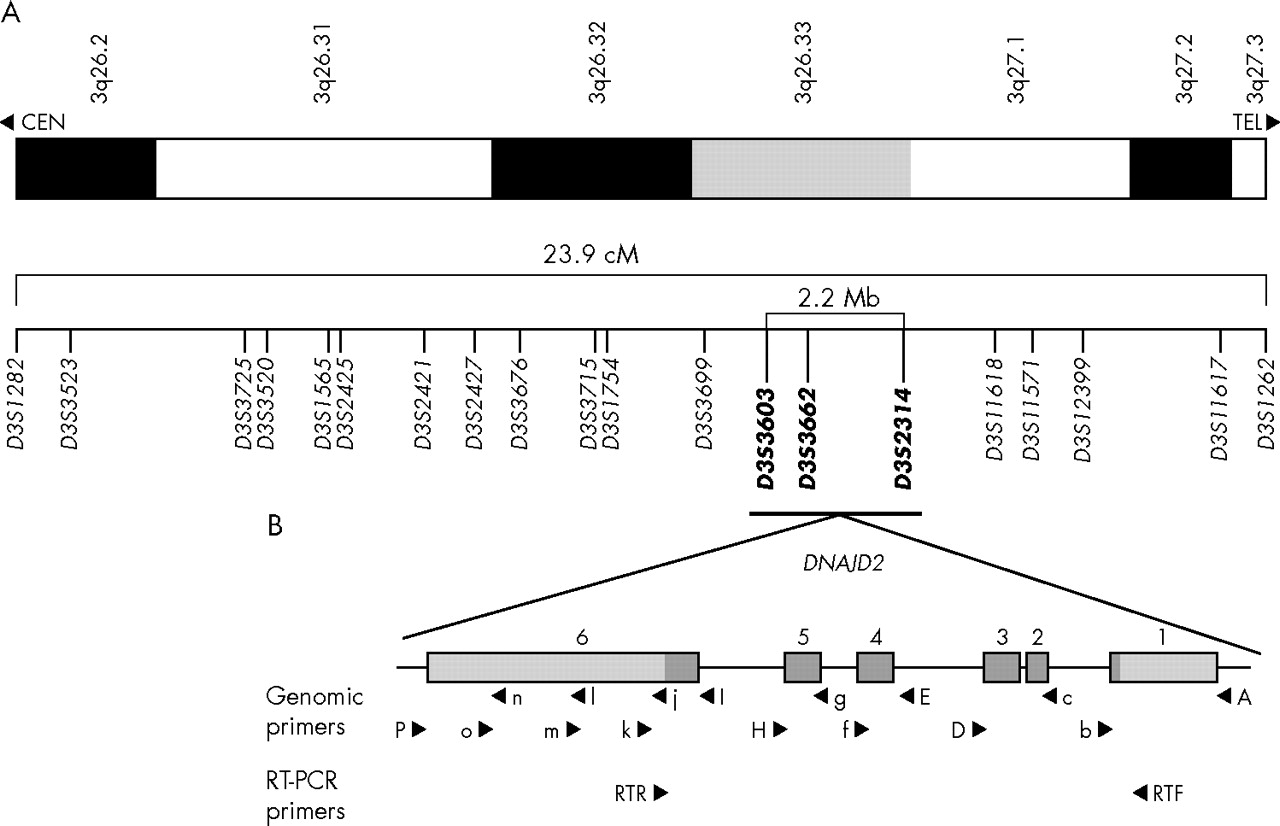
Map of the chromosome 3q DCMA candidate region. (A) Refined map interval spanning 23.9 cM from 3q26.2-q27.3 between markers D3S1282 and D3S1262. The markers in bold represent the core disease associated region of approximately 2.2 Mb (see electronic database information section for uniSTS accession numbers). (B) An expanded map of the genomic organisation of the DNAJC19 gene. The six exons extend over 5.2 kb. The coding sequence is indicated in dark grey and the non-coding sequence is in light grey. The locations of primers used for mutation screening are indicated. Primers used for amplification of three genomic segments are indicated by capital letters. Additional primers used for sequencing are shown in lower case. Primers used for reverse transcriptase polymerase chain reaction analysis are also indicated.
Sequencing reactions were carried out at 60°C using ABI BigDye version 3.1 (Applied Biosystems) and the following primers: exon1, forward: primer A, sequence shown above; exon1, reverse (b): 5′-AGCTGAGGTTGAGGCCTGGG; exons 2 and 3, forward (c): 5′-ATGGAAGAATGGGCAAAATG; exons 2 and 3, reverse: primer D, sequence shown above; exon 4, forward: primer E, sequence shown above; exon 4, reverse (f): 5′-AAGGAGAGAAGGTCTTTCTT; exon 5, forward (g): 5′-GTCTTTAATTGGCCTTTATG; exon 5, reverse: primer H, sequence shown above; exon 6, forward-1: primer I, sequence shown above; exon 6, forward-2 (j): 5′-AAATGCCTCAGAGCTACAAT; exon 6, forward-3 (l): 5′-TCTTATTCGGTGGAAAGGCT; exon 6, forward-4 (n): 5′-TGGGATTACAGGCATGAGTA; exon 6, reverse-1 (k): 5′-ATCCCCAAATTTAAACAAGA; exon 6, reverse-2 (m): 5′-TCCAAGTTGCCAGGCCAGTT; exon 6, reverse-3 (o): 5′-AGAAATAGCCAACTGAAGGA; exon 6, reverse-4: primer P, sequence shown above.
The DNAJC19 IVS3-1 G→C mutation abolishes a StuI cut site. Controls were tested for the presence of this mutation by PCR amplification of DNAJC19 segment 2 (primers and conditions given above). PCR product (10 μl) was digested for two hours in 3 units of StuI enzyme (Roche) and run on 1.5% agarose. The normal G allele when cut gives 1160 bp and 138 bp bands. The mutant C allele remains uncut and is a 1298 bp band. The G/C heterozygote gives 1298 bp, 1160 bp, and 138 bp bands (data not shown).
Reverse transcriptase polymerase chain reaction and tissue expression
Reverse transcriptase polymerase chain reaction (RT-PCR) was carried out on total RNA isolated from fibroblasts using the Qiagen RNeasy midi kit (Qiagen Inc, Valencia, California, USA). DNAJC19 specific primers were designed to amplify a 525 bp cDNA product corresponding to the full length coding sequence and the 5′ and 3′ UTR boundaries: RT forward primer (RTF), 5′-GGTAAAGGCGTGCAGGT; RT reverse primer (RTR), 5′-AAAATTGTAGCTCTGAGGCATT. RT-PCR was done using the Superscript One-Step RT-PCR kit (Gibco) with an initial incubation at 46°C for 20 minutes followed by 38 cycles at an annealing temperature of 55°C. cDNA RT-PCR products were sequenced using the RTF and RTR primers. Gene expression studies used cDNA panels containing a number of fetal and adult tissue types (BD Clontech multiple tissue cDNA panels I and II, and fetal multiple tissue cDNA panel (Clontech Laboratories, Palo Alto, California, USA). The cDNA panels were amplified using Platinum Taq buffer (Invitrogen) and the above RTF and RTR primers with an annealing temperature of 60°C, and run on 1.5% agarose.
Informatics
Map positions of markers and positional candidate gene models were identified using the National Center for Biotechnology Information (NCBI) map viewer. All primers were designed using Oligo 6. Protein domain predictions were made using the conserved domain architecture retrieval tool (CDART), and similarity searches based on this domain architecture were carried out using the reverse position specific BLAST algorithm. Other predictions were made using proteomics tools available from the expert protein analysis system (ExPASy). Multiple alignments were constructed using ClustalW, and alignments were visualised using Genedoc. Uniform resource locators for these tools and GenBank identification numbers for aligned sequences are provided in the electronic database information section below.
RESULTS
Clinical histories
Clinical features of our 18 study patients are summarised in table 1.
Summary of the clinical features of dilated cardiomyopathy with ataxia syndrome
Eleven of these patients had either echocardiographic or pathological features at necropsy consistent with a diagnosis of DCM. The onset of the DCM was always before the age of three years, and over 70% of affected patients died from either progressive cardiac failure or sudden cardiac death. Some patients improved with standard medical treatment, and complete resolution of the DCM was seen in two patients, with normalisation of the echocardiogram, even following withdrawal of cardiac drug treatment. Long QT syndrome was often seen in the patients with DCM; however, it was also seen in those without clinical or echocardiographic evidence of DCM. Prenatal or postnatal growth failure, or both, was universally seen. All patients over the age of two years had a cerebellar syndrome with ataxia resulting in significant motor delays, although most patients ambulate independently. Male genital anomalies ranging from isolated cryptorchidism to severe perineal hypospadias were common and were secondary to testicular dysgenesis, based on the frequent findings of small atrophic testes and incomplete testosterone rise after human chorionic gonadotrophin stimulation. A few male patients also had Mullerian remnants and therefore both androgen and anti-Mullerian hormone synthesis was impaired, consistent with testicular dysgenesis. Additional features included optic atrophy, mild increases in hepatic enzymes with microvesicular hepatic steatosis, a normochromic microcytic anaemia, and mild to borderline non-progressive mental retardation.
Many patients were found to have significant increases in urine organic acids, particularly of 3-methylglutaconic (3MGC) and 3-methylglutaric acid (3MGA). Quantitative measurement of the 3MGC and 3MGA7 was carried out in affected patients, controls, and unaffected relatives. Affected patients consistently showed five- to 10-fold increases in both plasma and urine 3MGC and 3MGA (data not shown).
Genome scan
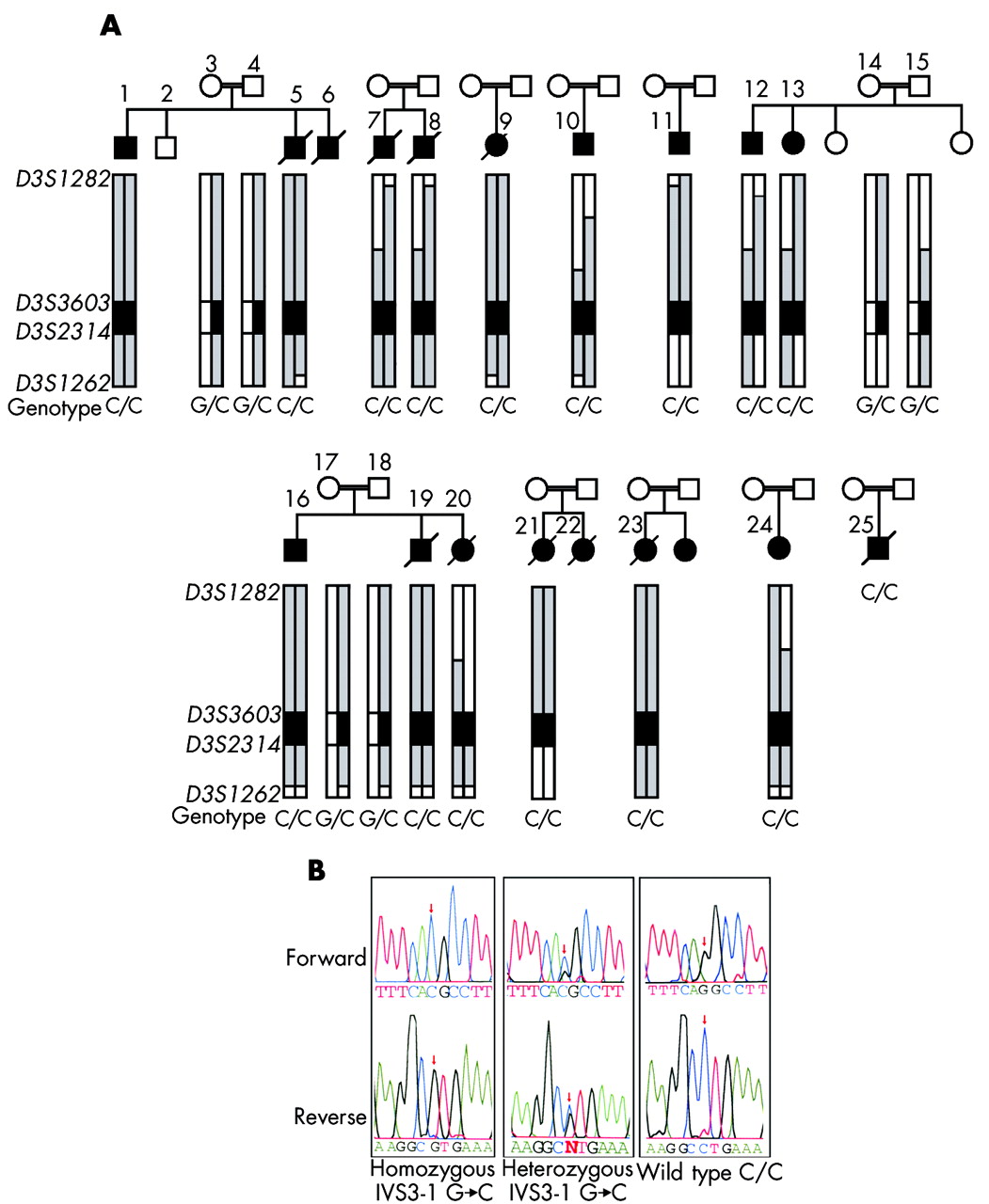
To identify the chromosomal region associated with the DCMA syndrome, we undertook a genome scan using a homozygosity mapping approach in five severely affected patients6 (fig 2A, individuals 1, 7, 9, 10, and 11). Following the initial scan, none of the markers was homozygous in all five patients. Homozygosity was suggested, however, at nine loci with either four of five, or three of five, patients homozygous for the same allele, and the non-homozygotes sharing the same allele on one chromosome (D2S2211, D2S325, D3S1565, D5S408, D12S1723, D12S310, D14S74, D22S420, and D22S315). Fine mapping around these candidate regions ruled out evidence of an ancestral homozygous haplotype in all but the chromosome 3 locus (D3S1565). Additional saturating markers enabled the identification of an extended disease associated haplotype of 23.9 cM on chromosome 3q26.2–q27.3, between markers D3S1282 and D3S1262 (fig 1A). An additional 10 affected patients, as well as any available unaffected family members, were genotyped, reducing this to a core region of 2.2 megabases between markers D3S3603 and D3S2314 on 3q26.33 (figs 1A and 2A).

The DNAJC19 IVS3-1G→C mutation is associated with a dilated cardiomyopathy syndrome in consanguineous Hutterite families. (A) Patients and families included in this study show an extended common haplotype from D3S1282 to D3S1262; alleles included in this haplotype are highlighted in grey with the core disease associated region highlighted in dark grey. The genotypes for the DNAJC19 IVS3-1G→C mutation are also shown. Patient numbers correspond with numbers in table 1. DNA was unavailable for patients 6 and 22; however, these patients were included in the study cohort, based on their clinical phenotypes. DNA for patient 25 was unavailable for haplotyping, but the clinical diagnosis was confirmed by genotyping for the DNAJC19 IVS3-1G→C mutation at a later date. (B) Electropherograms showing DNAJC19 IVS3-1G→C genotypes from an affected patient (C/C), an obligate carrier (G/C), and a wild type individual (G/G). These bases are reversed on the reverse sequence, as indicated below the figure.
Mutation screening
To identify the DCMA syndrome mutation in our patients, all predicted exons and splice junctions were bidirectionally sequenced for all 12 positional candidate genes in the minimal region. We identified a G→C transversion in the conserved AG splice acceptor site of intron 3 of the DNAJC19 gene (IVS3-1G→C) (fig 2B). DNAJC19 was previously uncharacterised, and comprises six exons spanning 5.2 kb of genomic sequence (fig 1B). All affected patients included in this study for whom DNA samples were available (n = 16) were homozygous for this mutation, while unaffected parents showed a pattern of inheritance of this mutation consistent with their carrier status of the affected haplotype (n = 8). No unaffected siblings were homozygous for this haplotye (n = 3) (fig 2A). The IVS3-1 G→C mutation eliminates a StuI restriction site. Mutation screening of 236 unaffected controls by StuI digest revealed only the normal G/G genotype (data not shown). No mutations were identified in any of the other 11 positional candidates in this region.
DNAJC19 gene expression
The DNAJC19 IVS3-1G→C mutation was predicted to prevent splicing of the exon 4 coding sequence into the DNAJC19 mRNA (Δex4). To confirm this effect, DNAJC19 specific RT-PCR analysis was carried out on total fibroblast RNA from an affected patient and an unaffected control. A single 445 bp product was detected in the affected patient, while two products—a dominant 525 bp band and a minor 445 bp band—were detected in the control sample (fig 3A).

Expression analysis of DNAJC19. (A) Reverse transcriptase polymerase chain reaction (RT-PCR) analysis of DNAJC19 using gene specific primers and RNA extracted from cultured fibroblasts from an affected patient and a non-Hutterite normal control. Two bands were amplified from the normal control, one of 525 base pairs (bp) corresponding to the full length cDNA, and a second less abundant band of 445 bp. In the affected patient, only the 445 bp band was detected. The predicted size of the product from a DNAJC19 transcript missing the exon 4 coding sequence is 445 bp. (B) Expression analysis of DNAJC19 was undertaken using gene specific primers and various normalised first strand cDNA preparations from fetal and adult tissues. Both cDNAs were found expressed in all tissues, with the full length cDNA more abundant in all tissues tested except for adult leucocytes. Tissue types tested: bn, adult brain; co, adult colon; fb, fetal brain; fh, fetal heart; fk, fetal kidney; fl, fetal lung; flv, fetal liver; fsk, fetal skeletal muscle; fsp, fetal spleen; ft, fetal thymus; h, adult heart; k, adult kidney; l, adult lung; lk, adult leucocytes; lv, adult liver; ov, adult ovary; p, adult prostate; pa, adult pancreas; pl, placenta; si, adult small intestine; sk, adult skeletal muscle; sp, adult spleen; ts, adult testis; ty, adult thymus. Each set was run with G3PDH, glyceraldehyde-3-phosphate dehydrogenase control; C, positive control; b, water blank. (C) To determine the identity of the cDNA products, the 445 bp cDNA from the affected patient, the 445 bp and 525 bp bands from adult leucocytes were sequenced. Both the cDNA from the affected individual, and the 445 bp band from leucocytes were found to have exons 3 and 5 spliced together and the absence of exon 4 coding sequence (Δex4). The 525 bp band corresponded to the full coding sequence of DNAJC19. The Δex4 cDNA detected in normal tissues and control are thought to be a normal splice variant.
To examine expression of the DNAJC19 gene in various tissue types, we PCR amplified the DNAJC19 cDNA, using multiple tissue first strand cDNA panels. The DNAJC19 transcript is ubiquitously expressed, as we detected a dominant 525 bp product corresponding to the full length DNAJC19 cDNA as well as a minor product of 445 bp in all tissue types tested (fig 3B). To confirm the identity of these products, we gel purified and sequenced the 445 bp band from the patient RT-PCR sample, as well as both the 445 bp and 525 bp bands from the adult leucocyte sample from the cDNA panel. The 445 bp products from the affected patient and the wild type adult leucocyte sample are missing coding sequence corresponding to exon 4 (Δex4), while the predominant 525 bp product from the leucocyte sample corresponds to the predicted full length mRNA sequence for the DNAJC19 gene (fig 3C).
Characterisation of DNAJC19
As DNAJC19 is a previously uncharacterised gene, various informatics analyses were applied to examine the consequence of the IVS3-1 G→C mutation on the DNAJC19 protein, and to identify any functional domains. The full length DNAJC19 gene encodes a predicted 116 amino acid protein with a molecular mass of 12.5 kDa. Translation of the Δex4 transcript is predicted to produce a truncated protein of 47 amino acids with a theoretical molecular weight of approximately 5 kDa. The first 43 residues encoded by exons 1–3 would be preserved; however, skipping of exon 4 produces a reading frame shift in the exon 5 coding sequence resulting in a predicted aberrant four amino acid substitution (PYCQ) followed by a termination codon.
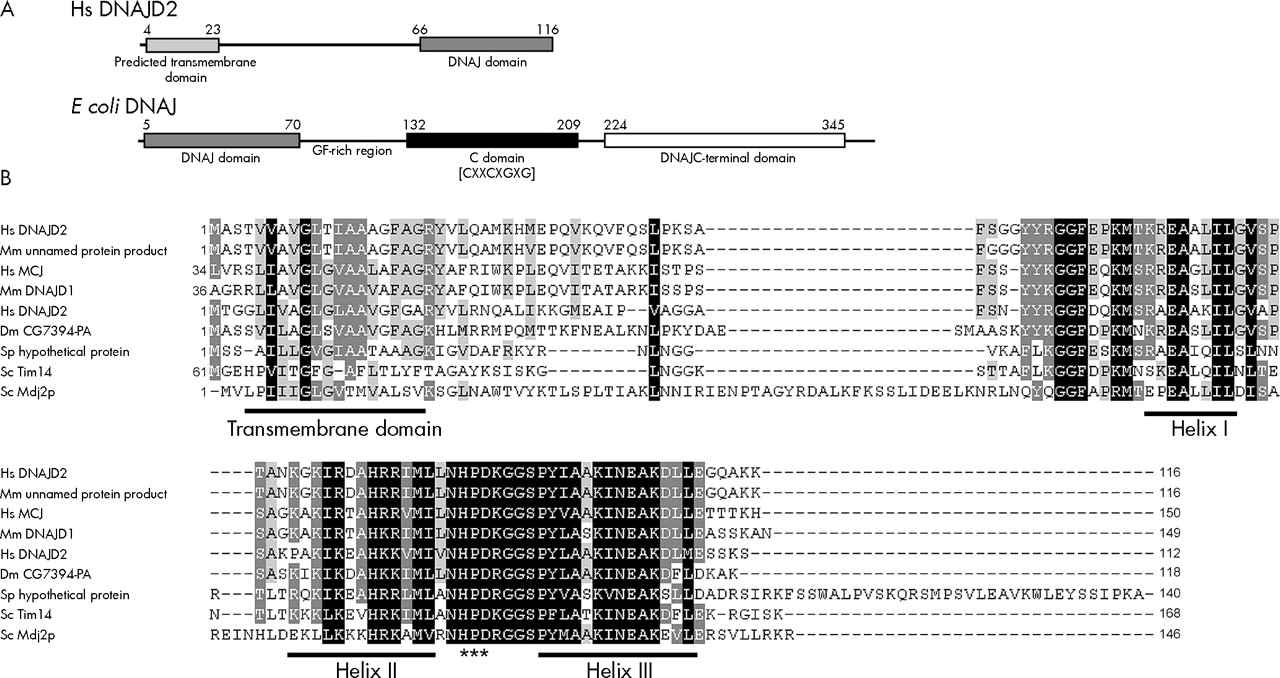
CDART analysis of the 116 amino acid DNAJC19 protein indicates the presence of a conserved DNAJ domain from residues 66 to 116 at the C-terminus of the protein. A single transmembrane segment was identified using the TMpred program at the N-terminus of the protein from residues 4 to 23 (fig 4A). In addition, the region directly following the transmembrane domain is predicted to form an amphipathic α helical structure that is rich in positively charged residues, resembling a mitochondrial targeting sequence. If translated, the truncated protein encoded by the Δex4 transcript would contain only a transmembrane domain, and would completely lack the DNAJ domain.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DNAJC19 has an unusual domain architecture with respect to other DNAJ proteins and similar proteins are found in various species. (A) Graphical representation of the domain structure of H sapiens DNAJC19 v the E coli DNAJ/HSP40 protein. The DNAJC19 protein contains only the DNAJ domain (pfam00226.11), which is located at the C-terminus following a predicted transmembrane domain. This is in contrast with the E coli DNAJ/HSP40 protein which is the classical example of a DNAJ protein. The DNAJ domain (pfam00226.11) is located at the N-terminus of the protein followed by a glycine/phenylalanine-rich linker domain, a cysteine-rich domain (pfam00684.11) containing four copies of a zinc-finger-like motif (CXXCXGXG), and a C-terminal domain (pfam01556.11) of unknown function. (B) ClustalW multiple sequence alignment of DNAJC19 homologues (see Electronic-Database Information section for GenBank identification numbers). The conserved helix structures of the DNAJ domain are underlined; the DNAJ domains of these proteins only contain three of the helices that make up the classical tetrahelical DNAJ domain. They all also contain the absolutely conserved HPD motif which is indicated by (***). Highlighted regions indicate conserved blocks of sequence. Residues highlighted in black indicate positions with highest degree of conservation and those highlighted in grey indicate less highly conserved positions. Gaps introduced to optimise the alignment are indicated by dashes (–). Alignment was adjusted in Genedoc based on previously published alignments.
Similarity searches were performed using rpsBLAST to identify proteins in other species that have similar domain architecture to DNAJC19. A number of similar proteins were identified that contained a highly conserved DNAJ domain at the C-terminus and a predicted transmembrane domain positioned towards the N-terminus, a selection of these are shown in figure 4B.
DISCUSSION
DCMA syndrome is a novel autosomal recessive disorder that shares some clinical similarity with the X linked Barth syndrome and the other 3-methylglutaconic acidurias. The three defined classes of 3-methylglutaconic aciduria—3-methylglutaconyl-CoA hydratase deficiency (type I), Barth syndrome (type II), and Costeff optic atrophy syndrome (type III)—are all caused by defects in known or predicted mitochondrial proteins.2–5 In addition, raised 3-methylglutaconic acid is a common finding in respiratory chain disorders. The major clinical and biochemical features of DCMA syndrome are consistent with a mitochondrial cytopathy; however, this degree of testicular dysgenesis has to our knowledge never been reported in a mitochondrial disorder. The autosomal recessive pattern of inheritance, and the unique combination of clinical features in DCMA syndrome, distinguishes it from the other 3-methylglutaconic acidurias and suggests that DCMA syndrome is a fifth type of 3-methylglutaconic aciduria.
To date, DCMA syndrome has only been seen in patients from the Dariusleut Hutterite population. It is possible that additional DCMA patients may exist outside this population, potentially in European ancestral populations from which the Hutterites descended. If this is the case, these patients may have been misclassified within the type IV 3-methylglutaconic aciduria category for lack of a better diagnosis, as cases of dilated cardiomyopathy not attributed to Barth syndrome have been included in this category.
Using a homozygosity mapping approach,6 we identified an ancestral haplotype on chromosome 3q26.2–3q27.3 segregating with DCMA syndrome. Mutation analysis of positional candidate genes identified a splice site mutation in a previously uncharacterised gene, DNAJC19, which encodes a mitochondrial DNAJ domain-containing protein.8 This mutation was predicted to cause aberrant splicing and result in the loss of the full length DNAJC19 transcript. RT-PCR analysis confirmed the splice defect (Δex4), and in affected patients homozygous for the IVS3-1G→C mutation splicing is completely abnormal and a full length DNAJC19 transcript is not produced. The presence of the Δex4 transcript in wild type tissues in addition to the full length transcript may indicate that alternative splicing is common at this site.
Proteins containing the DNAJ domain are typically involved in molecular chaperone systems of the Hsp70/Hsp40 type. These chaperone systems aid in folding and assembly of newly synthesised proteins, as well as preventing abnormal folding and aggregation of proteins during stress conditions. The Hsp70 protein binds unfolded hydrophobic domains of the substrate protein, while the Hsp40 co-chaperone aids in loading the substrate protein onto Hsp70 and stimulates Hsp70 binding activity through the DNAJ domain.9 The domain architecture of the DNAJC19 protein is unusual among members of the DNAJ protein family, in that the DNAJ domain is positioned at the C-terminus of the protein following a single transmembrane domain (fig 4A).10 This is in contrast with E coli DNAJ, the classical example of a DNAJ co-chaperone, where the DNAJ domain is located at the N-terminus, and is followed by other conserved domains involved in its other functions (fig 4A).9,10 In addition, the classical DNAJ proteins are soluble, whereas the predicted transmembrane domain of DNAJC19 suggests that it may be membrane bound. Similarity searches based on this domain architecture indicate that proteins similar to DNAJC19 are present in various species having a highly conserved C-terminal DNAJ domain and an upstream transmembrane domain (fig 4B). Two of these—the yeast Tim14 and Mdj2p proteins—are components of the mitochondrial inner membrane.11–14
The role of Tim14 has recently been elucidated in yeast, where it is a component of the TIM23 inner membrane translocase complex and is an essential protein required for cell viability.12–14 The TIM23 complex facilitates the import of mitochondrial presequence containing proteins through the inner membrane. Tim14 is an essential subunit of the TIM23 complex, functioning with mitochondrial Hsp70, and the Tim1615,16 and Zim1717,18 proteins as a component of the import motor subcomplex. This import motor is an adaptation of the Hsp70/Hsp40 chaperone system using a membrane bound DNAJ domain to stimulate binding of mitochondrial Hsp70 (mtHsp70) to the incoming peptide chain at its point of entry. This is thought to aid in import through the TIM23 translocase in two ways: first, ATP driven conformational changes in mtHsp70 upon substrate binding are thought to pull the incoming peptide into the matrix; and second, binding of mtHsp70 to the unfolded chain prevents backsliding through the Tim23 import pore.19 This system couples the chaperone function of mtHsp70 with import, aiding in proper folding and assembly of newly imported matrix proteins.
The functions of the other DNAJC19-like proteins are as yet unknown, and it is unclear whether DNAJC19 is in fact the human orthologue of yeast Tim14. The yeast Mdj2p protein is a known component of the inner mitochondrial matrix; however, a defined function has yet to be identified for it. What is known is that Mdj2p is not an essential protein for viability, and it can partially compensate for the loss of Mdj1p, the major mitochondrial hsp40 homologue in yeast.11 A second human protein, methylation controlled J protein (MCJ), is also included within this group.20 A differential pattern of expression has been noted for MCJ in wild type cell lines dependent on the methylation status of a CpG island in the first intron of the MCJ gene.21,22 Differences in expression of MCJ have been noted in ovarian cancers and may be associated with the cellular response to chemotherapeutic drugs.20,22 A proteomic screen of the mitochondria of human cardiac myocytes identified DNAJC19 as a component of the mitochondrial proteome; however, MCJ was not identified in this screen.8 Like DNAJC19, MCJ shares sequence similarity with the yeast Tim14 and Mdj2p proteins. The differential pattern of expression of MCJ, however, argues against a housekeeping-type role as would be expected for a Tim14 orthologue. The identification of DNAJC19 in the mitochondrial proteome, along with its role in the DCMA syndrome phenotype, are congruent with the DNAJC19 protein being orthologous with the yeast Tim14 or Mdj2 proteins.
The similarity of DNAJC19 with the yeast Tim14 protein suggests that the phenotype of DCMA syndrome might result from the defective import and assembly of presequence containing mitochondrial proteins through the TIM23 translocase pathway. Defects in mitochondrial import have been reported previously to cause a progressive neurodegenerative condition, Mohr-Tranebjaerg syndrome, which includes sensorineural deafness, vision loss, mental retardation, and a movement disorder.23 Mohr-Tranebjaerg syndrome is caused by mutations in DDP1,24 the orthologue of yeast Tim8, which appears to be important in facilitating the transfer of Tim23, the core component of the TIM23 inner membrane translocase complex, from the translocase of the outer membrane (TOM) through the intermembrane space to the TIM22 translocase.19,25 The phenotype of Mohr-Tranebjaerg syndrome is therefore thought to result from abnormal assembly of the TIM23 transporter. Mohr-Tranebjaerg syndrome is the only known genetic disorder involving a defect in the mitochondrial protein import system.
Over 99% of mitochondrial proteins in humans are nuclear encoded.19 Thus the mechanisms for transfer and assembly of the cytoplasmically synthesised precursor proteins into or across both mitochondrial membranes are of critical importance. The potential association of DCMA syndrome with mitochondrial protein import defects suggests that this may be an important cause of mitochondrial cytopathies. Mechanisms for protein transport and assembly in the mitochondria continue to be elucidated, largely on the basis of work in the yeast S cerevisiae. The relevance of these studies to our understanding of the pathogenic mechanisms underlying human mitochondrial diseases, however, remains poorly understood and further development of this area will benefit not only from the identification of additional human diseases but also from the development of mammalian model systems based on the studies already completed in simple eukaryotes. The present discovery of the association of DCMA syndrome with a mutation in the DNAJC19 gene will help to expand our insight into the role of mitochondrial import and trafficking in human cytopathies.
ELECTRONIC DATABASE INFORMATION
Entrez Genome Map Viewer, cytogenetic, physical and genetic maps of human chromosome 3 (http://www.ncbi.nlm.nih.gov/mapview/maps.cgi?taxid=9606&chr=3)
Oligo 6 primer design software (http://www.oligo.net)
Entrez UniSTS entries for markers making up ancestral DCMA syndrome haplotype (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unists): D3S1282 [68930], D3S3523 [57600], D3S3725 [73565], D3S3520 [17983], D3S1565 [63165], D3S2425 [82628], D3S2421 [13287], D3S2427 [37149], D3S3676 [30955], D3S3715 [74051], D3S1754 [9710], D3S3699 [43474], D3S3603 [30527], D3S3662 [78965], D3S2314 [15487], D3S1618 [72781], D3S1571 [8133], D3S2399 [10726], D3S1617 [39046], D3S1262 [53008]).
Entrez gene entries for positional candidates in the region D3S3603 to D3S2314 (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene): LOC131054 similar to RalA binding protein 1 (RalBP1) [131054], TTC14 tetratricopeptide repeat domain 14 [151613], hypothetical protein DKFZp434A128 [339829], LOC389178 similar to RING finger protein 13 [389178], LOC402151 similar to β-actin [402151], FLYWCH-type zinc finger 1-like [391594], LOC391595 similar to 60S ribosomal protein L32 [391595], fragile X mental retardation, autosomal homologue 1 [8087], DNAJ (Hsp40) homologue, subfamily D, member 2 [131118], SOX2 overlapping transcript (non-coding RNA) [347689], SOX2 SRY(sex determining region Y)-box 2 [6657], LOC401102 similar to zinc finger, CCHC domain containing 10 [401102].
Acknowledgments
We thank the Dariusleut Hutterite community, especially the individuals with this syndrome and their families, and the referring physicians for their participation and cooperation in this study. We also thank the staff of the Alberta Children’s Hospital Molecular Diagnostic Laboratory for their technical expertise. Special thanks to Dr Kym Boycott and Jayda Miller for their contributions. We acknowledge support from the Alberta Children’s Hospital Research Foundation, The Garrod Association and the Canadian Institutes of Health Research Training Program in Genetics, Child Health and Development.
REFERENCES
Footnotes
-
↵* These two authors contributed equally to this work.
-
Published Online First 3 August 2005
-
Conflicts of interest: none declared