Article Text

Abstract
Background: Peutz-Jeghers syndrome (PJS) is a rare, autosomal dominant cancer predisposition syndrome characterised by oro-facial pigmentation and hamartomatous polyposis of the gastrointestinal tract. A causal germline mutation in STK11 can be identified in 30% to 80% of PJS patients.
Methods: Here we report the comprehensive mutational analysis of STK11 in 38 PJS probands applying conventional PCR based mutation detection methods and the recently introduced MLPA (multiplex ligation dependent probe amplification) technique developed for the identification of exonic deletions/duplications.
Results: Nineteen of 38 probands (50%) had detectable point mutations or small scale deletions/insertions and six probands (16%) had genomic deletions encompassing one or more STK11 exons.
Conclusions: These findings demonstrate that exonic STK11 deletions are a common cause of PJS and provide a strong rationale for conducting a primary screen for such mutations in patients.
- MLPA, multiplex ligation dependent probe amplification
- PJS, Peutz-Jeghers syndrome
- MLPA
- multiplex ligation dependent probe amplification
- mutation
- Peutz-Jeghers syndrome
- STK11
Statistics from Altmetric.com
Peutz-Jeghers syndrome (PJS; MIM 175200) is a rare, autosomal dominant cancer predisposition syndrome characterised by oro-facial pigmentation and hamartomatous polyposis of the gastrointestinal tract.1 Depending on the patient population studied and analytical technique employed, a causative germline mutation in STK11 can be identified in between 30% and 80% of PJS patients.2–4 Failure to identify mutations in all PJS patients coupled with the report of a possible second disease locus has fuelled considerable debate as to whether allelic heterogeneity or genetic heterogeneity is responsible for PJS in which no STK11 mutation can be detected.5
Current screening methodologies for identifying STK11 mutations in PJS are typically PCR based, using heteroduplex methods, denaturing high performance liquid chromatography, and/or direct sequencing of the nine coding exons of the gene, together with whole fluorescent in situ hybridisation with a single STK11 probe or long range PCR. While these techniques can detect whole gene deletions and small scale intra-exonic deletions/insertions and point mutations, they are rarely capable of identifying deletions or duplications that encompass one or more exons. Such deletions are increasingly being recognised as being the mechanism of gene mutation in a number of cancer predisposition syndromes, such as hereditary breast-ovarian cancer syndrome, hereditary non-polyposis colorectal cancer, and Fanconi anaemia group A.6–8 Hence, the prevalence of STK11 mutations in PJS may have been underestimated in previously reported studies.
Several methods have been used to identify exonic deletions and duplication, including Southern blotting, long range PCR, quantitative fluorescent PCR, real time PCR, multiplex amplification and probe hybridisation, and multiplex ligation dependent probe amplification (MLPA). Of these, MLPA has been shown to be a highly sensitive method for detecting copy number changes in genomic DNA sequences and to be readily adaptable for high throughput screening.9 The technique relies on the ligation and subsequent PCR amplification of two adjacently hybridising probes using fluorescently labelled universal oligonucleotides complementary to synthetic sequence tags present on every probe. Each probe is designed to ensure a spectrum of uniquely sized PCR products is generated that can be individually quantified by electrophoretic analysis.
Here we report on the spectrum and prevalence of the different types of STK11 mutations in a series of 38 probands with PJS, specifically focusing on the possible contribution of exonic deletions to disease susceptibility. Analyses were restricted to British residents and clinical phenotype was not a selection criterion. In all cases the diagnosis of PJS was based on established clinical and pathological criteria.1 To screen for exonic deletions and duplications we made use of the STK11 MLPA assay kit, which has only recently become available (P101, MRC-Holland, Amsterdam, The Netherlands; www.mrc-holland.com). DNA was extracted from EDTA-venous blood samples using a standard salting-out procedure. Written consent was obtained from all participants and the study was conducted with local ethical approval in accordance with the tenets of the Helsinki declaration.
The 38 probands consisted of 15 males and 23 females (table 1). A family history of PJS had been documented in 21 (55%) of the probands. Using a combination of conformation sensitive gel electrophoresis, denaturing high performance liquid chromatography, and direct sequencing we identified germline STK11 mutations in 19 of the 38 (50%) probands. Details of these analyses and PCR primers have been previously reported.4 Nucleotide changes identified were coded according to the published sequence of STK11 (GenBank accession numbers: exon 1, AF032984; exons 2–8, AF032985; exon 9, AF032986) and mutations according to the Human Genome Variation Society (http://www.hgvs.org/mutnomen/examplesAA.html). The mutations detected in this phase of the analysis consisted of four nonsense mutations, six deletions, and two insertions predicted to lead to truncation of the expressed protein, four missense mutations, and three splice site mutations.
Clinical details and STK11 mutation status of the 38 PJS probands studied
A search for exonic rearrangements was conducted by MLPA in the 19 PJS probands in whom no germline STK11 mutation could be identified. This analysis was performed in accordance with the manufacturer’s protocol (MRC-Holland). The STK11 P101 assay is based on 28 probe sets: 13 that hybridise to the 10 exons and the 5′ untranslated region of STK11, and three that map 0.6 mb telomeric, 0.9 mb telomeric, and 10 mb centromeric to the gene; 12 probes serve as controls that hybridise to distal regions of the genome. PCR products were detected and data were analysed using an ABI 3100 semi-automated genetic analyser in conjunction with Genotyper software (version 3.7; Applied Biosystems, Foster City, CA, USA). For each sample analysed ratios of test peak area and control peak area to the mean control peak area were computed. Ratios were compared to mean ratios obtained following analysis of 19 control DNA samples from healthy unrelated individuals in order to normalise data between samples and probes to generate dosage quotients. Results were deemed usable if dosage quotients for each control peak per sample were between 0.8 and 1.2. Deletions and duplications were defined as two or more contiguous probes with dosage quotients of ⩽0.7 and ⩾1.3, respectively, in accordance with published recommendations.10 Figure 1 shows representative chromatograms obtained by MLPA analysis of the STK11 gene.
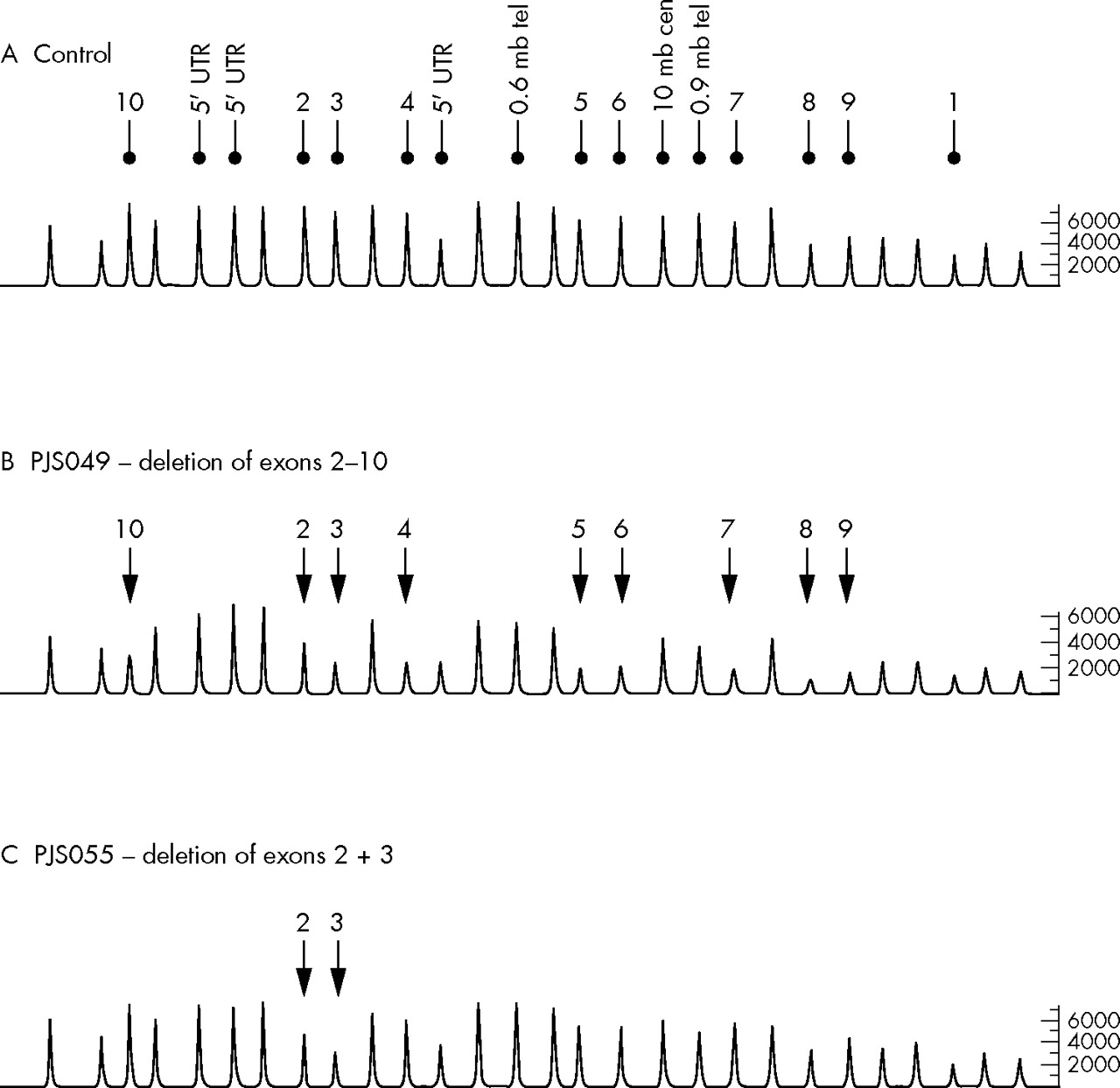
Representative chromatograms from MLPA analysis of STK11. (A) Control individual with peaks corresponding to exonic and 5′ UTR probes; probes hybridising to flanking regions of STK11 are indicated. All other peaks correspond to control probe signals. (B) Chromatogram from PJS patient PJS 049 showing the relative reduction in peak area of probes hybridising to exons 2–10 of STK11. (C) Chromatogram from PJS patient PJS 055 showing the relative reduction in peak area of probes hybridising to exons 2 and 3 of STK11.
MLPA analysis showed that six of the 19 PJS probands were carriers of exonic deletions (table 1): two unrelated cases with deletion of exon 1 and the 5′ furthest upstream probe (c.1−?_76+?del), one with deletion of the 5′ upstream sequence and exon 1 (c.1−?_76+?del), one with deletion of exons 2 and 3 (c.333−?_424+?del), one with deletion of exons 2–10 (c333−?_1302+?del), and one with deletion of exons 8 and 9 (c.1082−?_1183+?del). Figure 2 shows a representative ideogram of the mutations identified. For two of these cases (PJS 049: exons 2–10 deletion and PJS 062: exon 1 and 5′ upstream deletion) DNA from an additional family member was available, and in both cases an identical peak profile was observed in affected relatives. It has been suggested that single exon deletions should be confirmed by other methods to exclude false positive results from MLPA analysis,8 as reduced dosage quotients could be a consequence of point mutations in the probe hybridisation sequence thereby reducing the probe’s binding efficiency. It is, however, highly unlikely, that the deletions of multiple contiguous exons identified in this study represent false positives.8 All deletions identified in this report consist of multiple probe deletions and are, therefore, unlikely to represent false positive observations. Furthermore, two deletions were confirmed in affected family members and no reductions in test probe dosage quotients were observed in any of the 19 control samples tested. Neither of the two relatives evaluated had a personal history of malignancy.

{kind=link}
{kind=link}
Ideogram of STK11 deletions detected in PJS patients. Thick bars indicate extent of deleted sequence; thin bars indicate largest possible extent of deletion.
All mutations causing PJS reported to date, including those reported in this manuscript, result in either loss of STK11 kinase activity or disruption of STK11 regulatory regions.11 While large scale deletions of STK11 have previously been reported in PJS including deletion of approximately 250 kb encompassing the entire STK11 gene,12 these appear to be rare. To date most screens for large deletions in STK11 have been conducted using long range PCR. This has generally been based on amplification of exons 3–8, as an amplicon in excess of 20 kb is required to encompass the entire gene. It is apparent from this study that such analyses are inadequate as none of the exonic deletions reported in this study can be detected by long range PCR.
The findings of this study show that germline exonic deletions of STK11 are not a rare cause of PJS: 16% (6/39) of the entire cohort of PJS patients (95% confidence limits: 6% to 31%), and 32% of patients in whom mutations had not been previously identified (95% confidence limits: 13% to 57%), are readily detectable by MLPA analysis. These findings provide a strong rationale for screening for exonic deletions in STK11 in all PJS patients prior to conducting a conventional screen for point mutations and small deletions/insertions.
Given that various relationships between genotype and phenotype have been reported in PJS,13,14 it is probable that patients harbouring exonic deletions may have different manifestations to carriers of missense mutations. The number of patients analysed in our study is, however, too small for such relationships to be realistically examined.
The notion of genetic heterogeneity in PJS is founded on the observation that mutations in STK11 cannot always be identified in PJS patients, and evidence of genetic linkage of a PJS phenotype to 19q14 in a single family.5 Data from other mutational analyses of germline susceptibility suggest that the sensitivity of PCR based mutation detection methods is ∼70%. Hence, if the number of patients having PCR detectable mutations is adjusted accordingly, then in our study point mutations and small scale deletions/insertions may account for up to 86% of PJS (upper 95% confidence limit). Coupled with the relatively high prevalence of exonic STK11 deletions as a mechanism of disease susceptibility and the existence of cryptic splice sites as a cause of PJS15 means that almost all cases of the syndrome can be accounted for by STK11 mutations, thereby bringing into question the existence of locus heterogeneity.
Acknowledgments
We are grateful to the patients who participated in this study.
REFERENCES
Footnotes
-
This work was supported by grants from Cancer Research UK
-
Conflict of interest: none declared