Article Text

Abstract
Mental retardation is more common in males than females in the population, assumed to be due to mutations on the X chromosome. The prevalence of the 24 genes identified to date is low and less common than expansions in FMR1, which cause Fragile X syndrome. Systematic screening of all other X linked genes in X linked families with mental retardation is currently not feasible in a clinical setting. The phenotypes of genes causing syndromic and non-syndromic mental retardation (NLGN3, NLGN4, RPS6KA3(RSK2), OPHN1, ATRX, SLC6A8, ARX, SYN1, AGTR2, MECP2, PQBP1, SMCX, and SLC16A2) are first discussed, as these may be the focus of more targeted mutation analysis. Secondly, the relative prevalence of genes causing only non-syndromic mental retardation (IL1RAPL1, TM4SF2, ZNF41, FTSJ1, DLG3, FACL4, PAK3, ARHGEF6, FMR2, and GDI) is summarised. Thirdly, the problem of recurrence risk where a molecular genetics diagnosis has not been made and what proportion of the male excess of mental retardation is due to monogenic disorders of the X chromosome are discussed.
- mental retardation
- recurrence risks
- X chromosome
- X linked
- X linked mental retardation
- XLMR
Statistics from Altmetric.com
In 1938 Lionel Penrose first observed that more males than females in the population are mentally retarded in a survey and classification of those in institutional care and their relatives.1 The ratio of males to females was 1.25:1. This figure has been substantiated by numerous subsequent studies in the USA, Canada, Australia, and Europe and all agree with the observation of an approximately 30% excess of males being affected with mental retardation.2–6
The definition of mental retardation requires there to be significant sub-average general intellectual functioning (criterion A) that is accompanied by limitations in adaptive functioning in at least two of the following skill areas: communication, self care, home living, social/interpersonal skills, use of community resources, self-direction, functional academic skills, work, leisure, health, and safety (criterion B). The onset must also occur before 18 years of age (criterion C). General intellectual functioning is defined by the intelligence quotient (IQ). Adaptive functioning refers to how effectively individuals cope with common life demands. Although these observations are less objective measures and rely on information gathered from independent sources, for example, teacher evaluation and educational, developmental, and medical history, they are extremely useful in assessing children. In the UK, the ICD-10 Classification of Mental and Behavioural Disorders7 is used, while in the USA the DSM-IV diagnostic classification, which is similar to the WHO classification, is used.8
IQ across the population is normally distributed with the mean set at 100 and an IQ of <70 classified as mental retardation. Mild mental retardation is defined as an IQ of 50–70, moderate as an IQ of 35–49, severe as an IQ of 20–34, and profound as an IQ of <20. Approximately 2–3% of the population have mild to moderate intellectual disability and 0.5–1% of the population have moderate to severe mental retardation.
The publication in 1943 of the study by Martin and Bell9 subsequently led to the identification of the first single gene defect where the phenotype was predominantly mental retardation, Fragile X syndrome.10
In 1991 Kerr et al identified families where no clinical features other than mental retardation were observed and where Fragile X syndrome was not the cause of disease. This phenomenon was termed non-specific X linked mental retardation (XLMR).11 This then led to the classification of families with XLMR where a family was given an individual MRX number if linkage was performed and a LOD score >2.0 was obtained. The term MRX25 or MRX11 therefore has a precise meaning. To date MRX1 to MRX81 are recorded as individual families with XLMR, some of whom now have precise mutations identified in the literature (http://xlmr.interfree.it/5_non.htm).
DIAGNOSIS OF XLMR
The clinical diagnosis of XLMR is usually a diagnosis of exclusion of other causes of developmental delay in a male (table 1).12 Only rarely does a new family present with sufficient affected males for a confident clinical diagnosis of XLMR to be made. All patients where XLMR is suspected should have the benefit of contemporary karyotype analysis at >550 banded resolution, as unbalanced autosomal translocations from balanced carriers can be misclassified as X linked if no male-to-male transmission is observed. Similarly, with the advent of subtelomeric analysis approximately 3–4% of familial mental retardation will be found to be due to submicroscopic telomeric deletions.13–16 Mutation analysis for Fragile X syndrome is also essential.
Investigation of a male child with possible XLMR based on Shevell et al12
Having excluded a karyotype abnormality and Fragile X syndrome by seeking an expansion in FMR1, 23 X linked genes remain where mutations have been described that result in either syndromic or non-syndromic mental retardation (fig 1). The decision as to which gene(s) to analyse depends on the identification of additional clinical features that could categorise the condition as syndromic (table 2) and the relative prevalence of the gene abnormality in the study population.
Additional clinical features found in syndromic XLMR
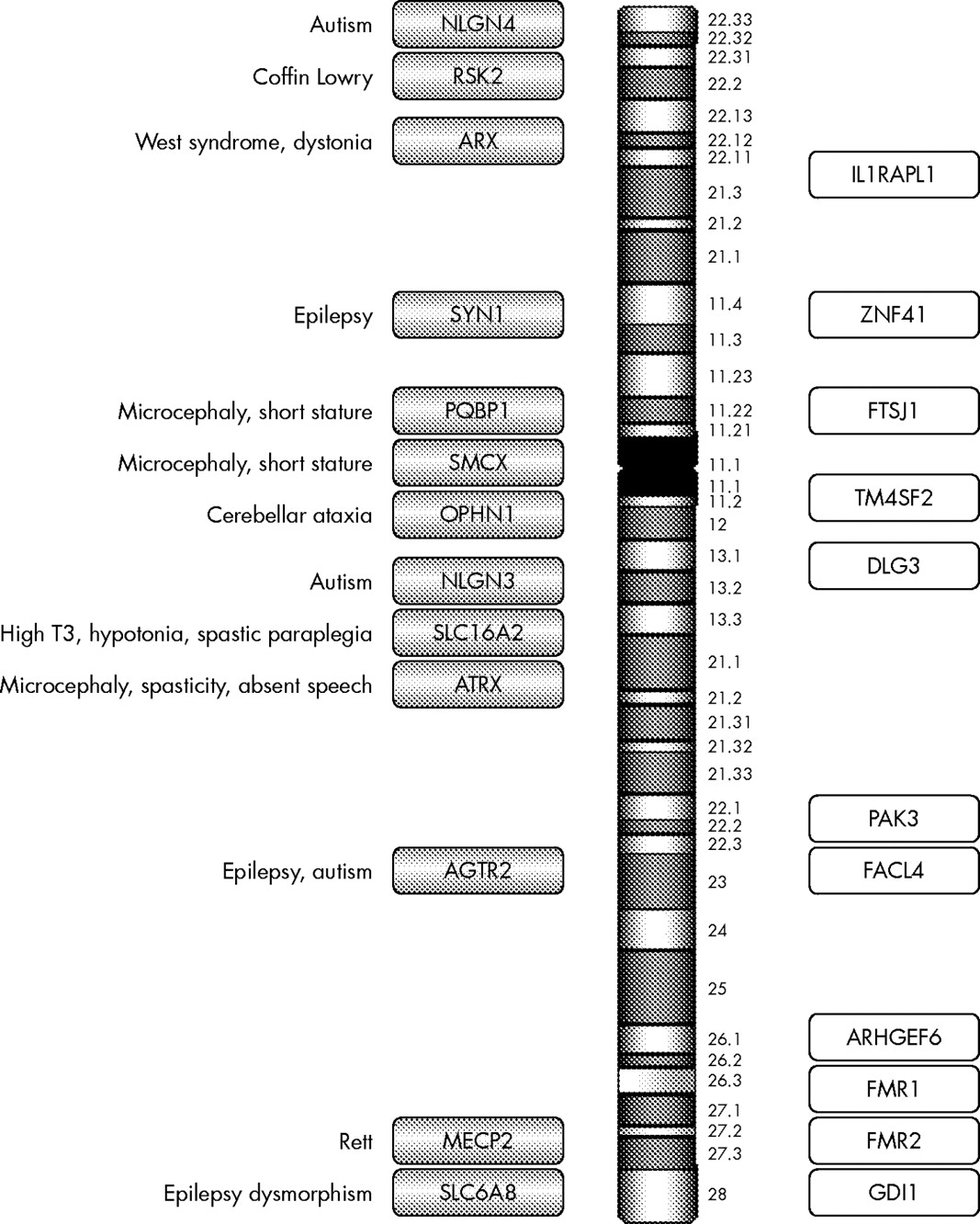
{kind=link}
Summary of genes on the X chromosome reported to cause XLMR. Shaded bars are syndromic XLMR genes; open bars are non-syndromic XLMR genes.
In this review a number of genes, the clinical features associated with the gene abnormality, and the prevalence of the disease gene will be discussed. Genes that are associated only with a syndrome are not discussed, for example L1CAM, PLP1, and DCX, as the review is limited to those genes where both non-syndromic and syndromic phenotypes have been described, due to limitations of space. The original separation of genes causing syndromic disease from those causing non-syndromic mental retardation was useful in the early days of identifying new genes that cause mental retardation. Increasingly, this divide is becoming blurred and somewhat arbitrary as the range of phenotypes associated with any one gene, for example ARX (see below), is becoming increasingly varied. Nevertheless, this distinction still has some limited merit when categorising genes associated with mental retardation.
SYNDROMIC XLMR
Autism
A mutation was identified in NLGN3 (neuroligin 3; OMIM 300366) and NLGN4 (neuroligin 4, X linked; OMIM 300427) in two brother pairs with severe mental retardation and autism.17 Since then a further family has been described, but mutations have not been identified in any large cohort of autistic children to date, suggesting that abnormalities in this gene are a rare cause of autism. In addition, all cases have been associated with severe mental retardation.18–20 Routine testing is of unproven utility to date.
Coffin Lowry
Mutations in RPS6KA3 (ribosomal protein S6 kinase, 90 kDa polypeptide 3; OMIM 300075), previously known as RSK2, are associated with Coffin Lowry syndrome.21 Short stature, distinctive facies with a prominent forehead and coarse facies, hypertelorism, prominent lips, large soft hands with thickened tapering fingers, hypotonia, hyperextensibility, and skeletal changes are characteristic.22 A single family with non-syndromic mental retardation has been reported with mutations in this gene, but recently mutations have been identified in three further families who did not meet the diagnostic criteria for Coffin Lowry syndrome (Raymond et al, unpublished data).23 This suggests that mutations in RPS6KA3 will prove to be a more common cause of XLMR and should be considered where the phenotype has some similarities. Testing therefore may yield further families.
Cerebellar ataxia
Families with mutations in OPHN1 (oligophrenin 1; OMIM 300127) were initially described as having a non-syndromic mental retardation phenotype, but on re-evaluation the affected males were found to have significant reduction in the size of the cerebellum. None of the families presented with significant ataxia or cerebellar signs clinically and the subtle cerebellar phenotype was only revealed on closer investigation. Obligate females also have reduced cerebellar size and this condition is now regarded as a syndrome.24,25 The prevalence of mutations in this gene is low, however systematic screening of this gene has not been performed in any large cohorts of patients with cerebellar hypoplasia or non-syndromic mental retardation. To date mutations have been described in two families, a patient with a translocation and a singleton with a similar phenotype to that of the familial cases.24,25 Testing in non-syndromic mental retardation alone is of unproven utility, but screening this gene in families with X linked cerebellar hypoplasia may be considered.
ATRX
X linked alpha thalassaemia was initially thought to be clinically homogenous, but mutation analysis of ATRX (alpha thalassaemia, mental retardation syndrome X linked; OMIM 300032) has found that the following conditions are all allelic: Juberg-Marsidi, Chudley-Lowry, Smith-Fineman-Myers, Carpenter-Waziri, Holmes-Gang, and Martinez. The phenotype is usually associated with severe mental retardation, commonly with absent speech, microcephaly, hypotonia, spasticity or seizures, and growth retardation with midface hypoplasia and skeletal abnormalities. A single large family has been reported with non-syndromic mental retardation alone where the proband did not have the characteristic facial features and profound intellectual disability associated with ATRX syndrome. Other affected members of the family did have the characteristic phenotype, suggesting that abnormalities of this gene show some intra-familial variation.26 Testing for this gene abnormality initially by screening for the presence of HbH bodies is certainly valuable where there is a syndromic phenotype, but routine screening of this gene in non-syndromic mental retardation is not useful.
Epilepsy
Mental retardation in combination with epilepsy is relatively common, which means that the list of differential diagnoses remains long in cases that present with these two features. However, mutations in SLC6A8 (solute carrier family 6 (neurotransmitter transporter, creatine), member 8; OMIM 300036) are usually associated with epilepsy, severe mental retardation, and autistic spectrum behavioural problems with particular deficits in expressive speech and language often resulting in absent speech.27 Recently, a systematic screen of 288 families with mental retardation and either with proven X linked inheritance or having two or more affected male family members revealed mutations in 6/288 (2.1%) families, suggesting that mutations in this gene are a relatively common cause of mental retardation, although still 10 times less frequent than Fragile X syndrome in familial cases.28,29 The clinical features of these six families were not described in detail, so the presence or absence of epilepsy as a diagnostic criterion is not entirely clear. Patients with this condition have altered creatine/creatinine ratios and reduced creatine uptake. In future, the detection of this condition using biochemical assays of plasma and urine will be an invaluable screen and mutation analysis will then be used as confirmation of disease in affected individuals.30–32
The identification of ARX (aristaless related homeobox; OMIM 300382) as a cause of West syndrome, mental retardation and either hypsarrythmia, myoclonic epilepsy, dystonia (Partington syndrome), lissencephaly, and abnormal genitalia or mental retardation alone has altered the previous somewhat rigid delineation of conditions as syndromic or non-syndromic, as the same mutation within this gene can lead to a wide variety of phenotypes.33,34 Intracerebral cysts have also now been reported.35 Problems associated with mood including aggression or depression were also a feature in some families. Within families where XLMR is highly suspected, the prevalence of mutations in this gene is relatively high at 9/136 (6.6%), but systematic screening of a larger cohort of smaller possible X linked families has revealed no mutations (0/151) and in further screening of affected singletons the prevalence is low (2/1501).36–38 Testing is useful in syndromic mental retardation and in familial cases of non-syndromic mental retardation but not in singleton cases.
A truncating mutation in SYN1 (synapsin 1; OMIM 313440) has been reported in a single family with mental retardation and epilepsy.39 This is not a common cause of mental retardation as more than 300 families with X linked or possible XLMR have now been screened and no new mutations have been identified to date (Raymond et al, unpublished data).
Mutations in AGTR2 (angiotensin II receptor, type 2; OMIM 300034) have been described in 10 patients to date, but the same mutation p.G21V found in three patients appears to be a rare polymorphism and unlikely to be disease causing.40–42 Severe mental retardation associated with epilepsy was present in the other cases reported with likely pathological mutations and two families had autistic behaviour. Testing is of unproven value to date.
MECP2
The clinical spectrum seen in males with mutations in MECP2 (methyl CpG binding protein 2 (Rett syndrome); OMIM 300005) include neonatal encephalopathy, Angelman syndrome, Rett syndrome, severe mental retardation with or without progressive spasticity, and manic depression as in PPM-X (mental retardation, psychosis, pyramidal signs, and macroorchidism X syndrome). Orrico et al reported a family where a mother had mild intellectual problems, a daughter had classic Rett syndrome, and four affected boys had severe mental retardation.43 All family members have a missense mutation A140V in MECP2, suggesting that mutations in MECP2 may be a common cause of mental retardation in males. Two further families were then reported, one with progressive spasticity and the other with PPM-X syndrome, where Q406X and A140V, respectively, were found to be the causative mutations.44,45 This stimulated screening of male patients with severe mental retardation for mutations in MECP2. Many sequence changes have been identified, but few are disease causing as this gene is highly polymorphic. Recent cohort studies have identified one pathological mutation in almost 1000 samples (1/475 in a European consortium study, 0/300 in the Cambridge GOLD study cohort, and 0/>200 samples referred for testing of males with mental retardation to Wessex Clinical Genetics Laboratory, UK).46 Mutations in MECP2 are rare causes of non-syndromic mental retardation and sequence analysis should not be routinely offered but remains invaluable in the diagnosis of Rett syndrome and related disorders with a high diagnostic yield.
SHORT STATURE AND MICROCEPHALY
Mutations in PQBP1 (polyglutamine binding protein 1; OMIM 300463) have been published as a cause for XLMR, but all patients described to date have a range of syndromic features. Microcephaly, short stature, and mental retardation are common features, together with a variety of mid-line defects including anal atresia, situs inversus, congenital heart disease, cleft palate, ocular coloboma, and small testes. One patient had spastic paraplegia. The phenotypes in Renpenning syndrome, cerebropalatocardiac (Hamel) syndrome, and Sutherland Hann syndrome are similar and mutations in PQBP1 have also been identified in these conditions, suggesting they are allelic.47–49 No non-syndromic mental retardation patients have yet been described, but screening patients with the above clinical features would be useful.
Mutations in SMCX (Smcy homolog, X linked (mouse); OMIM 314690), previously known as JARID1C, are also associated with short stature and microcephaly.50 Of the seven families described, only one family has a normal head circumference. Other frequent clinical features of this syndrome are small testes, prognathism or micrognathia, strabismus, myopia, facial hypotonia, progressive spastic paraplegia, epilepsy, and aggressive behaviour. Only one family had non-syndromic mental retardation and was relatively mildly affected compared to the others.50
HIGH TRIIODOTHYRONINE CONCENTRATIONS (T3)
Mutations in SLC16A2 (solute carrier family 16 (monocarboxylic acid transporter), member 2; OMIM 300095), also known as MCT8, a thyroid hormone transporter gene, were first reported in five unrelated boys with severe mental retardation and high triiodothyronine T3 concentrations.51 Two of the boys had partial deletions of the gene with a 24 kb deletion that encompassed exon 1 and a 2.4 kb deletion which resulted in a deletion of exon 3 and exon 4. The other three boys had missense mutations and a nonsense mutation.51 Subsequently, two further families were reported with abnormal triiodothyronine (T3) levels, global developmental delay, central hypotonia, spastic paraplegia, dystonic movements, rotary nystagmus, and impaired hearing and gaze.52 Children with Allan-Herndon-Dudley syndrome have a similar phenotype to that of the families reported by Dumitrescu et al and six large families have all been found to carry mutations, five missense and one 3 bp pair deletion.53 These patients were all subsequently found to have associated abnormal T3 levels although the neonatal Guthrie screens for hypothyroidism were normal. Abnormalities in this gene appear to be relatively common and suggest that in the diagnosis of profound mental retardation with neurological features detailed and continued surveillance of thyroid function tests may be helpful. This will aid the early identification of families who have mutations in this gene.
In the differential diagnosis of X linked spastic paraplegia (SMCX), SCL16A2 should be considered together with L1CAM (OMIM 308840), PLP1 (OMIM 300401), MECP2, and ARX.
NON-SYNDROMIC MENTAL RETARDATION GENES
Nine genes have been identified where the clinical feature of the families is mental retardation alone: IL1RAPL1, TM4SF2, ZNF41, FTSJ1, DLG3, FACL4, PAK3, ARHGEF6, FMR2, and GDI. To date, no detailed comparative prevalence studies have been published for abnormalities in these genes. The prevalence of Fragile X syndrome in affected sib pairs and X linked families is approximately 12/45 (27%), although this figure predates molecular genetic analysis and is likely to be an overestimate.29,54 The prevalence of each of the non-syndromic genes is 1–2% in selected research samples where at least two males are affected in the family pedigree.
IL1RAPL1 (interleukin 1 receptor accessory protein-like 1; OMIM 300206) was first identified as a candidate gene for mental retardation after the finding of deletions in families with mental retardation, adrenal hypoplasia, Duchenne muscular dystrophy, and glycerol kinase deficiency. Initially, a mutation was identified in 1/20 small XLMR families screened and no mutations were found in five large X linked families.55 Since then a complex rearrangement of this gene has been described, but no new mutations have been found.56
A translocation disrupting TM4SF2 (OMIM 300096), now known as TSPAN7 (tetraspanin 7), identified this gene as a potential cause of mental retardation. Mutations in this gene were also identified in 2/33 small families and 0/3 large families, but no further mutations have been reported since.57 More recently, the significance of the missense mutation p.P172H in one of the families has been questioned, although it has been reported in another case of a singleton with mild to moderate mental retardation but without family follow-up studies being carried out.58,59
Disruption of ZNF41 (zinc finger protein 41; OMIM 314995) in a child with mental retardation and a balanced X autosome translocation identified this as a candidate gene. Screening of a panel of 210 families with XLMR identified one missense and one splice site mutation which are likely to be pathological.60
Mutations in FTSJ1 (Fts J homolog (Escherichia coli); OMIM 300499) have been found in 2/219 small X linked families and 2/30 linked families. Three mutations affect splicing and one is a missense mutation.61,62
Four truncating mutations in DLG3 (discs, large homolog 3 (neuroendocrine-dlg, Drosophila); OMIM 300189) have been identified in a cohort of 328 families with XLMR.63 All the affected males in the families had moderate to severe mental retardation while female carriers were usually of normal intellect. X inactivation studies showed no skewing of lymphocytes in obligate female carriers.63
Mutations in FACL4 (renamed ACSL4, acyl-CoA synthetase long-chain family member 4; OMIM 300157) have been reported. These are two missense and one splice site mutation that reduce the enzymatic activity.64,65 The gene was originally localised by characterising genomic deletions in patients with Alport’s syndrome and mental retardation.66,67
Since the identification of a truncating mutation in PAK3 (p21(CDKN1A) activated kinase 3; OMIM 300142), two further missense mutations have been described.68–70 Only a few families have been screened to date. The initial study screened 18 families, all of whom had positive linkage data that mapped the gene abnormality in the family to Xq21, but no systematic prevalence data are available for this gene.
Disruption of ARHGEF6 (Rac/Cdc42 guanine nucleotide exchange factor (GEF) 6; OMIM 300267) in a balanced translocation patient and the identification of a single intronic IVS1-11T>C mutation in 1/119 mentally retarded patients have been described to date.71
FMR2 was identified by characterising the genomic structure around the folate sensitive fragile site, FRAXE.72,73 The official name for this gene is AFF2 (AF4/FMR2 family, member 2; OMIM 309548). Two unrelated boys with mental retardation had submicroscopic deletions in this region and facilitated the localisation of the gene. Subsequently, two families were identified, one of whom was also found to have FRAXA. The penetrance of FMR2 is variable and the phenotype can be mild or borderline mental retardation. Currently most diagnostic laboratories offer a PCR based screen for expansions in this gene. Specific Southern blot analysis can then be performed if the diagnosis is suspected. The prevalence is rare compared to Fragile X syndrome and interpretation of results is sometimes difficult.74–77
Three mutations in GDI1 (GDP dissociation inhibitor 1; OMIM 300104) have been characterised. Two out of five X linked families with linkage data mapping to Xq28 have mutations and 1/164 males with non-familial mental retardation have been screened and found to carry a mutation.78,79
Finally, Fragile X syndrome should be considered as a frequent cause of non-syndromic mental retardation as the classic phenotype of mental retardation, macrocephaly, frontal bossing, large ears, prominent mandible with prognathism, and enlarged testes is rarely seen. The prevalence of an expansion in the 5′CGG repeat of FMR1 (fragile X mental retardation 1; OMIM 309550) in the population in an unselected sample of mainly singletons is 1/3500–1/9000 and many of the affected individuals have a non-syndromic phenotype.80,81
RECURRENCE RISKS FOR MENTAL RETARDATION
Where a molecular genetic diagnosis has been made, accurate genetic advice can usually be given. Unfortunately, this is still rarely the case when a family presents in a clinic and recurrence risks need to be quantified. Much of the available data were published between 1971 and 198782–86 (table 3) and although the observations of recurrence risks of 2–14% are still valid, the quality of chromosome analysis, the advent of molecular genetic testing for Fragile X syndrome, and the improved clinical expertise in syndromic identification question the validity of some of these data.87 A recent population based study in Atlanta, USA discusses contemporary recurrence risks for developmental delay although the sample was relatively old by contemporary molecular and cytogenetic standards. The sample was based on children born to mothers between 1981 and 1991 and the total number of cases with a disability was 3685. Recurrence risks for isolated mental retardation were 8.4% if the first child had isolated mental retardation as compared with recurrence risks for cerebral palsy of 2.9–3.6%, hearing loss of 4.7–5.7%, and vision impairment of 5.3–6.9%. If the first child had mild mental retardation, recurrence risks for mild and severe mental retardation were 7.1% and 4.7%, respectively.88
Summary of published recurrence risks for mental retardation by sex of proband
Where detailed clinical assessment is possible and current molecular genetic and cytogenetic analysis is available, the calculations of Turner and Partington are an additional useful guide for calculating recurrence risks.89 These authors observed recurrence risks for mental retardation in the siblings of index cases referred to a genetics centre. Observed recurrences were 1:7.5 (11/83) for brother pairs and 1:20 (3/60) for sister pairs. These figures are comparable to those of Herbst and Miller from 1980 and those from the Colchester cohort collected by L Penrose and revisited by Morton et al.90,91 The calculated offspring risk to intellectually normal siblings of a single affected male were 1–2% for the offspring of a normal male sibling and 2–5% for the offspring of a sister. The figures include the risk of an undetected familial cryptic translocation and, for the sister, the estimated risk that disease in a singleton male brother is due to an X linked disease (∼25%). If there are two affected males in a family, the assumption is that ∼80% (the male excess) is due to X linked disease. The offspring risk to a normal brother remains the same as the risk of an undetected familial cryptic translocation, as above, whereas the offspring risk to an unaffected sister if there are two affected males is significantly higher at 10% to include the X linked disease risk. Although this guide is useful, the calculations are inevitably inaccurate as they are biased by ascertainment of families in a clinical genetics setting and assume that the majority of male sib pairs have X linked disease.
Mandel and Chelly have addressed the issue of whether the observed male excess of patients with mental retardation is due entirely to mutations in monogenic disease genes on the X chromosome or not.37 Observing the prevalence of a 24 bp expansion in ARX, they observed that 6.6% (9/136) of families with XLMR pedigrees but only 0.13% (2/1501) of singleton cases were found to carry this mutation. Based on this observation, they calculate that only ∼10% of the excess males observed are due to X linked genes. This observation does not alter the practical clinical recurrence risks we inform patients about, but suggests that the identification of the cause of mental retardation in some families, especially where a single generation is affected, will be even harder to elucidate. Accurate genetic counselling of those families where no mutation is identified will continue to be challenging in the future. The use and predictive value of predisposing alleles or polymorphisms in clinical practice is extremely limited and this situation is not likely to change. Furthermore, this suggests that genetic counselling should clearly distinguish families with an X linked pedigree from those where a single generation is affected and provide appropriate recurrence risks based on the probability of there being an X linked condition in the family.
To date more than 20 genes have been identified that cause XLMR. Estimates of the number of genes that remain to be identified vary considerably from 30 to 50.92–94 Until all the genes on the X chromosome have been scrutinised in a large sample cohort, the exact number of XLMR genes will remain unknown, as will the prevalence and importance of each gene as a cause of human mental retardation. The future challenge is to understand the molecular genetic basis of the observed excess of mentally retarded males, discover the autosomal causes of mental retardation, and determine the biological basis of this disease in each gene abnormality identified.
In summary, Fragile X syndrome remains the most common XLMR gene discovered so far. Syndromic features should always be sought in possible XLMR as this can lead to molecular diagnosis. The discovery of the plethora of genes that cause a small proportion of non-syndromic XLMR has clinical value for those families where mutations are detected, but awaits the arrival of high throughput, cheap, and reliable sequence analysis methods that can be readily introduced to clinical service.
Acknowledgments
I would like to thank Professors G Turner, M Partington, and J Goodship for discussion of the clinical phenotype of the three families with novel mutations in RPS6KA3, and Dr G Woods for his helpful comments on the manuscript. I would like to thank the British Clinical Genetic Society for the invitation to present a review of X linked mental retardation in March 2005 which formed the basis of this article.
REFERENCES
Footnotes
-
Published Online First 23 August 2005
-
The GOLD study project is supported by the Wellcome Trust and is being carried out in collaboration with the Wellcome Sanger Institute.
-
Competing interests: none declared