Article Text

Abstract
Background: An inherited germline P53 mutation has been identified in cases of childhood adrenocortical carcinoma (ACT), a neoplasm with a high incidence in southern Brazil. The penetrance of ACT in carriers of the point mutation, which encodes an arginine-to-histidine substitution at codon 337 of TP53 (R337H), has not been determined.
Objective: To investigate the penetrance of childhood ACT in carriers of the R337H TP53 mutation.
Methods: The family histories of 30 kindreds of 41 southern Brazilian children with ACT were obtained. A PCR based assay was used to detect this P53 mutation in a large number of relatives of children with ACT. In all, 927 individuals were tested for the mutation, 232 from the non-carrier and 695 (including the 40 probands) from the carrier parental lines.
Results: 40 children with ACT carried the TP53 R337H mutation; the remaining child with ACT was not tested. There was no evidence of Li-Fraumeni syndrome in any of the kindreds; however, seven met the criteria for Li-Fraumeni-like syndrome. The carrier parental line was identified in each kindred. Of the 695 individuals tested in the carrier parental line, 240 (34.5%) were positive for the mutation, while none of the 232 individuals in the other parental line carried the mutation. The penetrance of ACT was 9.9% (95% confidence interval, 8.7% to 11.1%).
Conclusions: The TP53 R337H mutation dramatically increases predisposition to childhood ACT but not to other cancers, and explains the increased frequency of ACT observed in this geographic region.
- ACT, adrenocortical tumour
- LF, Li-Fraumeni syndrome
- MLE, maximum likelihood estimator
- PBL, peripheral blood lymphocytes
- TP53
- penetrance
- adrenocortical tumour
- Li-Fraumeni syndrome
- childhood adrenocortical carcinoma
Statistics from Altmetric.com
- ACT, adrenocortical tumour
- LF, Li-Fraumeni syndrome
- MLE, maximum likelihood estimator
- PBL, peripheral blood lymphocytes
In southern Brazil, the annual incidence of childhood adrenocortical tumour (ACT) is unusually high, ranging from 3.4 to 4.2 cases per million children-year,1 compared with a worldwide incidence at ages 0–14 years of only 0.3 per million children-year.2 Germline mutations in the DNA binding domain of the TP53 gene are associated with Li-Fraumeni (LF) syndrome, which is characterised by an increased risk of cancer, including paediatric ACT.3,4 Notably, in carriers of these TP53 mutations, ACT accounts for 3% of all tumours occurring at any age and for 10% of childhood tumours. Thus TP53 mutations appear to increase the predisposition of children to ACT. Although childhood ACT is frequently observed in families that have susceptibility to cancer, relatives of southern Brazilian children with ACT do not appear to have the cancer profiles of LF3 or LF-like syndrome,5 as has been reported previously.1 The Brazilian patients typically carry a unique TP53 mutation (R337H) in the oligomerisation domain of the protein.6 The aetiology of this mutation, which appears to be restricted to a specific region of Brazil, remains elusive. However, the loss of heterozygosity with retention of the mutant allele in tumour cells, the accumulation of TP53 protein in the nucleus, and the folding characteristics of the missense protein in vitro6,7 strongly support the role of the R337H mutation in tumorigenesis.
The R337H mutation, which encodes histidine instead of arginine at codon 337, disrupts a HhaI restriction endonuclease site. This genetic alteration allowed the development of a reliable screening method to detect carriers and establish the penetrance of ACT among carriers. Because tumour size is highly predictive of prognosis in children with ACT,8 close monitoring of children at risk may allow early intervention and improved outcome.
In this study, we sought to answer the following questions: What is the frequency of the R337H TP53 mutation among relatives of children with ACT? Is there evidence of the de novo emergence of this mutation? Is there any evidence that this TP53 mutation increases the risk of other cancers? What is the penetrance of childhood ACT in carriers of the R337H TP53 mutation? What is the cumulative incidence (risk) and age specific risk of childhood ACT among carriers?
METHODS
Subjects and their relatives
Of the 32 girls and nine boys with ACT, 40 were tested and found to carry the R337H TP53 mutation. The remaining child with ACT was not tested, although her four tested siblings were carriers of the mutation. Among the parents, 21 mothers and 16 fathers were carriers. Parents of one proband were not available for testing. The children were 0.7 to 10.8 years of age at the time of diagnosis (mean (SD), 3.18 (2.43) years; median 2.1 years). Complete pedigrees were constructed for the 30 kindreds with one (22 kindreds) or more (eight kindreds) cases of ACT. Relatives from both parental lines of each proband were invited to participate in the study and were interviewed for history of cancer. In all, 927 individuals were tested for the mutation, 232 from the non-carrier and 695 (including the 40 probands) from the carrier parental lines. On the basis of the pedigree, the number of additional carriers among the untested 281 individuals in the carrier parental line was estimated. The latter individuals were not tested because either they failed to reply or they declined the invitation to participate.
The study was approved by the ethics committee of the Hospital de Clinicas, Federal University of Paraná, the Brazilian national ethics committee, and the institutional review board of St Jude Children’s Research Hospital. Written informed consent was obtained from all probands’ parents and adult relatives and from the legal guardians of relatives younger than 18 years of age.
PCR assay
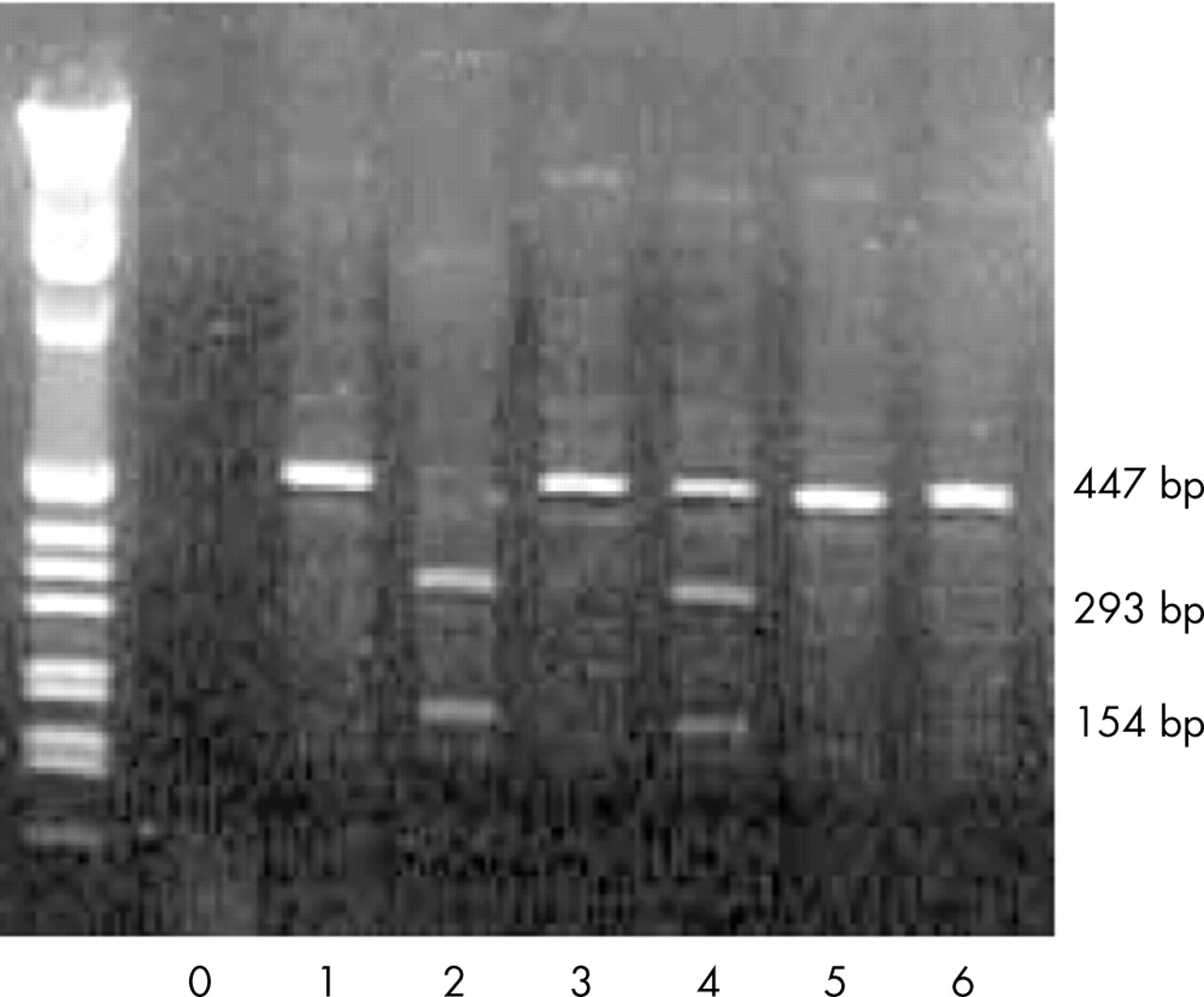
Genomic DNA from peripheral blood lymphocytes (PBL) and tumour cells was isolated and amplified by polymerase chain reaction (PCR) using standard protocols. Specifically, PCR was carried out in a 50 μl PCR buffer containing 2.5 U Taq polymerase (Perkin-Elmer, Norwalk, Connecticut, USA), 100 pmol of forward and reverse primers, 25 nmol each of deoxy-NTPs (Perkin-Elmer), and 100–200 ng genomic DNA. The PCR product corresponding to a 447 base pair (bp) segment of exon 10 was generated using the following primers: 5′-CTG AGG CAC AAG AAT CAC-3′ (forward) and 5′-TCC TAT GGC TTT CCA ACC-3′ (reverse). Samples were denatured at 95°C for 45 seconds and amplified by 30 cycles (annealing at 62°C for 45 seconds and primer extension at 72°C for 45 seconds) using a Perkin-Elmer thermal cycler. The amplified product (20 μl) was digested with HhaI, yielding 293 bp and 154 bp fragments only if the R337H point mutation was not present (fig 1). Digestion products were separated by electrophoresis in a 1.8% agarose gel prepared in Tris-borate-EDTA buffer containing ethidium bromide. The results of the PCR analysis were validated by sequencing the entire TP53 gene (22 patients) or exon 10 of the TP53 gene (five patients).

Polymerase chain reaction (PCR) amplified, HhaI-restricted DNA from control leucocytes (lane 2), a proband’s leucocytes (lane 4), and the same proband’s tumour (lane 6). Lane 0 shows a negative control (no DNA). Lanes 1, 3, and 5 are undigested PCR-amplified products from control (1), proband’s blood (3), and proband’s tumour (5). Lane 2 shows two bands (154 and 293 bp) representing both wild-type p53 alleles. Lane 4 shows 3 bands from a subject heterozygous for the R337H TP53 allele (HhaI cleaves wild-type P53 but not the R337H mutant). Lane 6 shows a single band from genomic tumour cell DNA that has undergone loss of heterozygosity.
Statistical analysis
The maximum likelihood estimation method for pedigree design proposed by Gail et al9 was modified to estimate penetrance. The complex pedigree structures and the number of untested individuals in our dataset made the summation over all possibilities of untested genotypes in Gail’s model impractical. However, the pedigree structures allowed us to derive the genotypes of certain untested individuals and simulate the genotypes of the remaining untested individuals in each pedigree. Thus likelihood data for the complete set of subjects (in which genotypes of untested individuals were either derived or simulated) were used to estimate penetrance. The following assumptions were made for deriving and simulating genotypes of untested individuals and for constructing the likelihood function:
-
there are no de novo mutations;
-
only one proband’s parent is a carrier;
-
for an untested individual, if one of their parents is a carrier, then the probability is 50% that this individual is a carrier;
-
kindreds are independent of each other;
-
within kindreds, observed phenotypes are conditionally independent given the individuals’ genotypes.
The following rules for deriving the genotypes of certain untested individuals were based on the pedigree structure of each kindred. First, if the proband’s parents were untested, then one was designated a carrier and the other a non-carrier; if one was tested, then the genotype of the other was considered opposite to that of the tested individual. Second, with respect to a carrier’s parents, set the parent who has parents in the parental carrier line of the pedigree to carrier, and recursively apply this rule to untested individuals in the upper generations. Third, set the other parent in each of these cases to non-carrier status. Penetrance estimators and confidence intervals were obtained by repeatedly simulating the genotypes of the remaining untested individuals according to the pedigree structure of each kindred and then applying maximum likelihood estimation along with the calculation of confidence intervals in each simulation trial. Genotype simulation was carried out using the following procedure: for each individual of unknown genotype in the generation being considered (generation 2, 3, 4, and/or 5), if one of the parents is a carrier (Aa), then the individual is set to carrier with 50% probability or non-carrier (AA) with 50% probability. If both parents are non-carriers, then the individual is set to non-carrier. This is a first order Markov Chain model10; the progeny’s genotype distribution given the ancestry genotypes is completely determined by parental genotypes. Finally, the penetrance was estimated by the following procedure: (1) derive the genotypes of certain untested individuals according to the rules described above; (2) simulate the genotypes of the remaining untested individuals according to the first order Markov Chain model; (3) apply maximum likelihood estimation to obtain a maximum likelihood estimator (MLE) and a 95% confidence interval (CI) for the penetrance; (4) repeat steps 2 and 3 1000 times. The final penetrance estimate is the average of the 1000 MLEs. The final 95% confidence interval is obtained by averaging the lower and upper ends of the 1000 confidence intervals.
The cumulative incidence (risk) of ACT among carriers under age 19 years was estimated using the non-parametric cumulative incidence function estimator of Kalbfleisch and Prentice.11 All carriers (except the probands), including those whose genotypes were derived or simulated, were assumed to have survived ACT-free for at least 19 years and were censored at 19 years of age. The genotypes of untested individuals were derived and simulated. Cumulative risk and risk were calculated during each simulation trial. The final estimates were computed by averaging the estimates across 1000 simulations.
RESULTS
Frequency of TP53 R337H mutation
In the carrier parental line, 695 individuals were tested. These included 40 children with ACT, 74 of their parents, and 16 grandparents. Fifty per cent of the parents and grandparents carried the R337H TP53 mutation. Thus there was no evidence of de novo mutation. In one kindred, a member born more than 100 years ago was assumed to have the mutation because both children were carriers (fig 2). Table 1 lists the characteristics of the 30 kindreds.
Characteristics of 30 kindreds from southern Brazil with one (n = 22) or multiple (n = 8) cases of adrenocortical tumour with the R337H TP53 mutation

Truncated pedigree of kindred 30 with two probands (V.4 and V.10). Families of individuals III.3 to III.17 are not shown. An individual in the first generation appears to be the oldest carrier of the mutation identified to date (born more than 100 years ago); this carrier was the parent of individuals II.1 and II.3, who were born 84 and 82 years ago, respectively. Square, male; circle, female; empty symbols, non-carriers; filled symbols, probands; half filled symbols, carriers of the R337H TP53 mutation; question marks within square or circle, untested individuals; diagonal line, deceased individuals; Roman numerals, generation numbers; Arabic numerals, case numbers.
All together, the R337H TP53 mutation was found in 240 of the 695 (34.5%) tested individuals in the carrier parental line. In the contralateral parental line, the mutation was not found in any of the 232 individuals tested. The frequency of the mutation was 38% (87/228) in the eight kindreds with more than one case of childhood ACT and 33% (153/467) in kindreds with only one case.
Cancer history in the carrier parental line
Thirty one cases of cancer were reported among first and second degree relatives of the probands. The germline mutation status of three of the four first degree relatives with cancer was known: all were carriers of the R337H TP53 mutation. The remaining first degree relative with cancer was assumed to have the R337H TP53 mutation because his sibling was known to be a carrier of the mutation. The mutation status of the other relatives with cancer was not known. The tumour types included breast (n = 5), stomach (n = 5), brain (n = 4), oesophagus (n = 3), prostate gland (n = 3), throat (n = 3), lung (n = 2), liver (n = 2), uterus (n = 1), bone (n = 1), axilla (n = 1; unknown tumour type), and abdomen (n = 1; unknown tumour type). Among the probands’ first degree relatives, only three of 37 parents and one of 86 siblings who carried the mutation had developed cancer; these individuals experienced tumours of the bone (mother, kindred 7), breast (mother, kindred 18), abdomen (tumour of unknown type; father, kindred 25), and brain (sister, kindred 25) (table 2). Twenty seven cases of cancer were reported among second degree relatives of the probands; nine cases occurred at age 45 years or less and 18 cases occurred after age 45 years. None of the kindreds had classical Li-Fraumeni syndrome.3 However, LF-like syndrome, as defined by Birch et al,5 was identified in seven of the 30 kindreds.
History of cancer in 30 kindreds of 41 probands with the R337H TP53 mutation
Penetrance of the R337H TP53 mutation
The risk of ACT among carriers of the mutation was calculated on the basis of the number of cases of ACT and the number of carriers of the mutation. Because the carrier frequency was 34.5% among individuals tested, 97 individuals—including one child with ACT—were expected to be carriers among the 281 family members who were not tested. The modification of Gail’s likelihood estimation method (detailed above) yielded an overall derived penetrance of 9.9% (95% CI, 8.7% to 11.1%). The penetrance derived by this method for kindreds with more than one case of ACT (12.5% (10.2% to 14.6%)) was greater than that for kindreds with only one case of ACT (8.5% (7.3% to 9.7%)).
Because ACT in these kindreds typically affects young children (mean (SD) age, 3.2 (2.4) years; range 0.7 to 10.8), we estimated the age specific risk and the cumulative risk of ACT for each year of the first 19 years of life (fig 3). There was clearly an increased risk between the ages of 1 and 2 years. The cumulative incidence reached a plateau at age 10 years. The derived penetrance of 9.9% (table 3) does not exactly match the cumulative risk of 11.3% at 12+ years (fig 3) because of the use of different statistical models, although the values are comparable.
Frequency of derived genotypes and estimate of penetrance

{kind=link}
{kind=link}
{kind=link}
Age specific and overall cumulative risk of adrenocortical tumour in carriers of the R337H P53 mutation.
DISCUSSION
We found the estimated (derived) penetrance of ACT among carriers of the R337H TP53 mutation to be 9.9% (95% CI, 8.7% to 11.1%). The higher estimated penetrance in some kindreds suggests that other still unknown factors may contribute to the tumorigenic potential of the mutation. The cumulative incidence of ACT differed according to age, peaking in the first two years. In this age group, the observed incidence of ACT was about three times that of the 8 to10 year old age group. The cumulative incidence reached a plateau at age 10 years in our cohort, although ACT has been reported in young adult carriers of the R337H TP53 mutation.12 These data suggest that approximately one of every 10 carriers of this TP53 mutation develops ACT. However, this figure was calculated on the basis of data from families in which at least one child had ACT and therefore should be interpreted with caution. The penetrance of ACT in carriers of DNA binding domain TP53 mutations has not yet been determined.
Interestingly, carriers of the R337H TP53 mutation do not show an increased risk of the cancers typically associated with DNA binding TP53 mutations. In the latter cases, there is a pervasive family history of cancer with a lifetime cancer penetrance of 73% for male carriers and virtually 100% for female carriers.13 Moreover, in carriers of DNA binding TP53 mutations, bone and soft tissue sarcomas and breast cancers predominate, while childhood ACT comprises only about 3% to 4% of tumours.14 In carriers of the R337H TP53 mutation, in contrast, we found childhood ACT to represent at least 57% (41/72) of all cancers, and no case of soft tissue sarcoma was recorded. These observations corroborate the narrow tissue specificity of certain TP53 mutations, in particular the R337H mutant.
It is not known whether the R337H TP53 germline mutation increases the predisposition to other malignancies during the course of the carrier’s life. In the carrier parental line, we observed several tumour types in second degree relatives more than 45 years of age. Because there is no population based tumour registry in this region of Brazil, it was impossible to determine whether the incidence of malignancies in carriers differed from that in the general population. Four malignant neoplasms were reported in first degree relatives under the age of 45 years. Whether the incidence of extra-adrenal tumours in first degree relatives (4/123; 3.25%) is higher than that in the general population remains to be determined. However, it is worth noting that the family history of extra-adrenal cancers was negative in first degree relatives in 26 of these 30 families. A more rigorous comparison of the frequency and types of cancer in carriers versus non-carriers of the mutation should elucidate whether the R337H TP53 germline mutation increases the predisposition to extra-adrenal cancers.
Several factors suggest that the R337H TP53 mutation is relevant to the high incidence of ACT in this Brazilian region. First, the R337H mutation was not found in 200 unselected paediatric patients seen at the endocrinology clinic of the Hospital de Clinicas in Curitiba6 or in 232 relatives in the non-carrier parental lines in our study group, whereas more than 95% of children with ACT in this region were carriers. Second, in every case of ACT associated with this germline mutation, loss of heterozygosity with retention of the R337H TP53 allele in tumour tissue was documented.6 Finally, the R337H TP53 mutation exerts a range of physical and biochemical effects not observed with wild-type TP53.7
We were unable to demonstrate any case of de novo mutation. In all families tested, one parent was consistently found to be a carrier of the mutation. In the few families in which both grandparents in the carrier lineage were available, one of each pair was a carrier. The oldest carrier tested was an 86 year old male. However, the structure of the pedigree (fig 2) provides compelling evidence that the mutation has existed for more than 100 years. The origin of the mutation and its role in increasing the carrier’s predisposition to paediatric ACT in a restricted geographical region of Brazil remain elusive. The mutation is not clustered in any single ethnic group or geographical location within the southern region of Brazil. Our previous study of a limited number of families showed no evidence of a single founder effect.6 Moreover, the incidence of ACT is not increased in most regions of Brazil, and the R337H TP53 mutation was not identified in any of 10 cases of paediatric ACT in north east Brazil (data not shown). It is possible that environmental factors were responsible for introducing this TP53 mutation into the genetic pool in this region. Another possibility is that the R337H TP53 occurs naturally at an extremely low frequency in certain populations but interacts with other, as yet undiscovered, specific genetic changes to increase the predisposition to paediatric adrenocortical carcinoma. Recently, Longui et al15 reported the presence of missense germline mutations and loss of heterozygosity of the inhibin α subunit gene in some cases of adrenal tumour in Brazilian children with the R337H TP53 mutation. Moreover, naturally occurring polymorphisms of TP53 have been shown to alter its function and increase predisposition to specific cancers.16 We are currently sequencing a large number of unrelated R337H TP53 alleles to determine whether they have polymorphisms that may be functionally relevant. In addition, we will determine the frequency of the R337H TP53 mutation in the southern Brazilian population. These data should provide insight into the molecular epidemiology of this tumour.
Paediatric patients are rarely screened for malignancies. In selected instances of inherited cancer susceptibility, however, screening is warranted. For example, the early detection and treatment of familial retinoblastoma cures nearly 100% of patients while preserving vision in the affected eye.17 Similarly, when detected at an early stage, ACT is almost always curable, whereas large or metastatic ACT carries a dismal prognosis.8 The availability of a reliable molecular marker to detect the R337H TP53 mutation allows the rapid identification of carriers in families that have a child with ACT. Once identified, carriers could be screened for early detection of ACT by imaging and endocrine studies. Whether early detection of ACT will reduce the mortality in these patients remains to be determined.
Acknowledgments
We wish to thank Kristen Shannon for reviewing the manuscript content, Dr Mary-Ellen Conley for technical assistance in developing the PCR screening tests, and Sharon Naron for editing the article.
Supported in part by Cancer Center Support grants CA-21765 and CA-71907 from the National Institutes of Health (US Department of Health and Human Services); a Center of Excellence grant from the State of Tennessee; the Conselho Nacional de Pesquisa (CNPq) of Brazil; Fundacao Araucaria, Curitiba, Parana, Brazil (01/2001), and the American Lebanese Syrian Associated Charities (ALSAC). RMP is the recipient of a PhD scholarship from CAPES (Brazil).
REFERENCES
Footnotes
-
Published Online First 20 July 2005
-
Conflicts of interest: none declared