Article Text

Abstract
Congenital hypothyroidism is the most common neonatal metabolic disorder and results in severe neurodevelopmental impairment and infertility if untreated. Congenital hypothyroidism is usually sporadic but up to 2% of thyroid dysgenesis is familial, and congenital hypothyroidism caused by organification defects is often recessively inherited. The candidate genes associated with this genetically heterogeneous disorder form two main groups: those causing thyroid gland dysgenesis and those causing dyshormonogenesis. Genes associated with thyroid gland dysgenesis include the TSH receptor in non-syndromic congenital hypothyroidism, and Gsα and the thyroid transcription factors (TTF-1, TTF-2, and Pax-8), associated with different complex syndromes that include congenital hypothyroidism. Among those causing dyshormonogenesis, the thyroid peroxidase and thyroglobulin genes were initially described, and more recently PDS (Pendred syndrome), NIS (sodium iodide symporter), and THOX2 (thyroid oxidase 2) gene defects. There is also early evidence for a third group of congenital hypothyroid conditions associated with iodothyronine transporter defects associated with severe neurological sequelae. This review focuses on the genetic aspects of primary congenital hypothyroidism.
- ERSD, endoplasmic reticulum storage disease
- NIS, sodium iodide symporter
- PHP, pseudohypoparathyroidism
- PPHP, pseudopseudo‐hypoparathyroidism
- PTH, parathyroid hormone
- TPO, thyroid peroxidase
- TRH, thyrotropin releasing hormone
- TSH, thyroid stimulating hormone (thyrotropin)
- congenital hypothyroidism
- candidate gene
Statistics from Altmetric.com
- ERSD, endoplasmic reticulum storage disease
- NIS, sodium iodide symporter
- PHP, pseudohypoparathyroidism
- PPHP, pseudopseudo‐hypoparathyroidism
- PTH, parathyroid hormone
- TPO, thyroid peroxidase
- TRH, thyrotropin releasing hormone
- TSH, thyroid stimulating hormone (thyrotropin)
Congenital hypothyroidism is detected at a rate of 1 in 3000 to 4000 live births, making it the most common congenital endocrine disorder.1 On a worldwide basis, hypothyroidism, including congenital forms, results most commonly from iodine deficiency. Otherwise congenital thyroid gland insufficiency results from developmental abnormalities at any level of the hypothalamic-pituitary-thyroid axis. Congenital hypothyroidism is most commonly caused by defects in thyroid development leading to thyroid dysgenesis (85%), which in turn consists of either thyroid agenesis (40% of all cases) or failure of the gland to descend normally during embryological development with or without ectopy (40%), or hypoplasia of a eutopic gland.2 The remaining cases are associated with either a goitre or a normal thyroid gland.3 Rarely, central (secondary) hypothyroidism may be caused by pituitary or hypothalamic disease leading to deficiency of thyrotropin (TSH) or thyrotropin releasing hormone (TRH), respectively, which will not be covered by this review.
In mammals the thyroid gland is the first glandular tissue to appear in development, arising from two regions of the endodermal pharynx which migrate and associate to become the characteristic bi-lobed structure.4 The median anlage, which eventually forms the follicular cells, arises from the midline of the anterior pharyngeal floor between the first and second branchial arches, being visible from days 16 to 17 of human gestation. At the same time two lateral anlagen (ultimobranchial bodies), which mainly become the parafollicular calcitonin secreting C cells but also contribute to a significant number of follicular cells,5 develop as caudal projections from the fourth or fifth pharyngeal pouches. The thyroid gland begins to trap iodide and therefore to secrete thyroid hormones only at 10 to 12 weeks of human gestation,4 with the transplacental passage of maternal thyroid hormones before this being important for fetal development.
The essential role of thyroid hormones in central nervous system (CNS) maturation has been clearly demonstrated,6 and congenital hypothyroidism is eminently treatable by thyroxine replacement. Screening for neonatal hypothyroidism has therefore been established on a worldwide basis, beginning in Canada in 1974 and in the United Kingdom in 1982. The development of infants with congenital hypothyroidism has been revolutionised with the institution of early and adequate treatment afforded by screening, thereby preventing intellectual impairment and infertility. This is witnessed by the normal growth and neurological development in affected children, and by a dramatic increase in IQ, from an average of less than 80 to the mean of the general population.7
Most cases of congenital hypothyroidism occur sporadically. However, dyshormonogenetic cases are often recessively inherited, and recent cohort analyses estimate that approximately 2% of cases with thyroid dysgenesis are familial.8 It is also noteworthy that congenital hypothyroidism is associated with an increased incidence of birth defects, with surveys in the United Kingdom reporting a non-thyroidal congenital anomaly rate of 7% and an increased prevalence of chromosomal anomalies (1.5%).9,10
The candidate genes associated with primary congenital hypothyroidism can be divided into two main groups: those causing thyroid gland dysgenesis and those associated with defects in the organification of iodide, leading to dyshormonogenesis. Genes associated with thyroid gland dysgenesis include those causing non-syndromic congenital hypothyroidism (TSH receptor) and those causing syndromic congenital hypothyroidism (TITF-1, TITF-2, PAX-8, and Gsα). Genes associated with dyshormonogenesis include those for thyroid peroxidase (TPO), thyroglobulin (TG), the sodium iodide symporter (NIS), pendrin (PDS), and most recently, thyroid oxidase 2 (THOX2). Recent evidence suggests a third group of congenital hypothyroid conditions associated with defects in the iodothyronine transporter, MCT8, where hypothyroidism is associated with severe neurological deficits.
In this review we will focus on the genetics of the primary congenital hypothyroidism. We will summarise the different phenotypes associated with the currently known genetic defects and outline informative investigative management.
THYROID DYSGENESIS AND NON-SYNDROMIC CONGENITAL HYPOTHYROIDISM
The TSH receptor gene (TSHR) encodes a transmembrane receptor present on the surface of follicular cells which mediates the effects of TSH secreted by the anterior pituitary and is critical for the development and function of the thyroid gland. It belongs to a subfamily of heptahelical G protein coupled receptors that have a common structure consisting of seven transmembrane segments, three extracellular and three intracellular loops, an extracellular amino terminal domain, and an intracytoplasmic carboxyl terminal tail (fig 1).11 The actions of TSH on the thyrocyte occur principally by receptor mediated activation of Gsα and subsequent generation of intracellular cyclic adenosine monophosphate (cAMP).12 The human TSHR gene is located on chromosome 14q31 and the extracellular domain of the receptor is encoded by nine exons, whereas the transmembrane and intracellular portions are encoded by a single large exon.13,14

Cartoon of the TSH receptor showing the positions of all the loss of function mutations reported to date. Missense mutations are shown in the circles, frameshift and deletion mutations are indicated by arrows, and splice site mutations are marked.
The initiation of expression of the TSHR on day 14 of mouse embryogenesis, at the onset of thyroid differentiation after completion of gland migration,15 suggests that alterations in TSH signalling pathways could result in defective thyroid development. The hyt/hyt mouse is homozygous for a loss of function mutation in the fourth transmembrane domain of the TSHR, resulting in hypothyroidism and thyroid hypoplasia postnatally.16
In contrast to mouse models, where an intact TSH-TSHR signalling pathway does not appear to be a prerequisite for the development of a normal sized thyroid gland in utero,17 this pathway is clearly important for the development of a normally sized fully differentiated gland in utero in humans.18 Homozygous or compound heterozygous mutations in the human TSHR gene resulting in TSH resistance were first described in 1995.19 Since then, additional loss of function TSHR mutations have been identified in other families showing variable TSH resistance because of reduced thyroid sensitivity to TSH (fig 1).20–32 In some of these families, compensated TSH resistance was associated with euthyroid hyperthyrotropinaemia and either a normal or a hypoplastic thyroid gland.19,20,27,29–31 Mild or borderline hypothyroidism, with greatly raised serum TSH associated with low normal or slightly low thyroid hormone levels and a normal thyroid gland, was seen in two families.21,22 At the other end of the spectrum, severe hypothyroidism associated with a hypoplastic thyroid gland or even apparent athyreosis has been reported.23–26,28,32
Inactivating mutations of the human TSHR are therefore associated with three phenotypes:
-
fully compensated TSH resistance to TSH;
-
partially compensated TSH resistance to TSH;
-
severe uncompensated resistance to TSH.
In common with other G protein coupled receptors, activating mutations have also been described in the TSHR, causing thyroid hyperfunctioning adenomas when somatic,33 or non-autoimmune congenital hyperthyroidism with germline abnormalities.34 It is of note that heterozygosity for a TSHR mutation (W546X) is increasingly being reported in families from a white background,20,21,32,35 with a rate of approximately 0.6% in a cohort of euthyroid Welsh subjects.35 These results have therefore raised the possibility that the mutation may contribute to thyroid dysfunction in that population, though this is very unlikely in the homozygous state.
There have also been reports of mildly raised TSH levels in individuals who are heterozygous for loss of function germline mutations in the TSHR.19,27,30–32 All have been reported to have normal sized eutopic thyroid glands on ultrasound scanning, except one who had thyroid hypoplasia.32 The clinical phenotype of these individuals is not readily distinguishable from those with compensated TSH resistance who are homozygous or compound heterozygous for mutations in the TSHR gene. Thus in families where non-autoimmune subclinical hypothyroidism appears to be inherited in an autosomal dominant manner, the possibility of a heterozygous mutation in the TSHR should be considered.
THYROID DYSGENESIS AND SYNDROMIC CONGENITAL HYPOTHYROIDISM
Evidence to date suggests that the development of the embryonic thyroid gland and its normal migration is dependent on the interplay between several transcription factors. In the thyroid gland, the target genes of these transcription factors include the thyroglobulin and thyroid peroxidase (TPO) genes. Transcription factors that have been studied in human congenital hypothyroidism include TTF-1, TTF-2, and the paired homeodomain factor Pax-8. In mice, the expression of TTF-1, TTF-2, and Pax-8 begins at the onset of thyroid migration on day 9.5 of gestation and these factors continue to be expressed throughout embryonic development.15,36,37 The onset of thyroid differentiation is heralded by the expression of TSHR, TPO, and TG on day 14.5 of mouse gestation.
TTF-2 is a transcription factor that is a member of the forkhead/winged helix domain protein family, many of which are key regulators of embryonic pattern formation and regional specification.36 It is a phosphoprotein that consists of an N terminal region, a highly conserved forkhead domain, an α helical polyalanine tract, and unique C terminal residues.38 Unlike some other transcription factors, such as HOX D13, polymorphism in the size of the polyalanine tract in TTF-2 has been shown not to affect its transcriptional function.38 The human gene is located in chromosome 9q22 and consists of a single exon.39 Animal studies have demonstrated the critical role of TTF-2 in thyroid embryonic development. Heterozygous Titf-2 knockout mice are euthyroid, with no visible phenotype, whereas homozygous null mice have cleft palate and thyroid dysgenesis, consisting of either thyroid agenesis or an ectopic sublingual gland, which is often lethal in the neonatal period.40 In mouse embryos, Titf-2 is known to be expressed not only in the thyroid gland but also in the craniopharyngeal ectoderm involved in palate formation and in Rathke’s pouch.36 More recently, its expression has also been demonstrated in pharyngeal endoderm derivatives, such as tongue, palate, epiglottis, and oesophagus, and in choanae and whisker follicle in the mouse and in human thyroid, hair follicle, and prepubertal testis.41,42
Two Welsh male siblings with a constellation of defects similar to those in the homozygous knockout mice (congenital hypothyroidism associated with thyroid agenesis and cleft palate)—with added features of spiky hair, bilateral choanal atresia, and hypoplastic bifid epiglottis43—were investigated for defects in human TITF-2 (also known as FKHL15 or FOXE1) and found to be homozygous for a missense mutation (A65V) within its highly conserved forkhead DNA binding domain.39 The mutant TTF-2 protein showed impaired DNA binding and loss of transcriptional function. This report represented the first description of a genetic cause for thyroid agenesis in humans. Subsequently, two siblings were studied from an unrelated consanguineous family, with a milder phenotype consisting of thyroid agenesis, cleft palate, and spiky hair, but with absence of choanal atresia and bifid epiglottis (fig 2).44 They were homozygous for a different missense mutation (S57N) in the forkhead domain of TTF-2, and the S57N mutant protein showed only partial loss of DNA binding and transcriptional activity in vitro, possibly explaining the incomplete clinical phenotype.

Patients with TITF-2 mutations, showing spiky hair, micrognathia, and hypertelorism (A), and cleft palate (B). (From: Castanet M, et al. A novel loss-of-function mutation in TTF-2 is associated with congenital hypothyroidism, thyroid agenesis, and cleft palate. Hum Mol Genet 2002;11:20521–9; reproduced by permission of Oxford University Press.)
TTF-1 is a homeobox transcription factor of the NK-2 gene family which is related to Drosophila NK-2/vnd (ventral nervous system defective).45 The homeobox gene superfamily encodes transcription regulatory proteins that act at critical points in development and ontogeny by sequence specific DNA binding, mediated by a structurally conserved homeodomain.46 TTF-1 has two independent transcriptional activation domains located at the amino terminal (N domain) and the carboxy terminal (C domain) regions with respect to the DNA binding homeodomain.47 The human locus is found on chromosome 14q13, and the gene (also known as NKX-2.1) comprises three exons encoding a 42 kDa protein.48
Initial evidence of a role for TTF-1 (also known as thyroid specific enhancer binding protein, T/EBP) in the aetiology of congenital hypothyroidism also came from a report of a mouse model in which the Titf-1 gene had been disrupted by homologous recombination.49 Heterozygous animals were initially described as having a normal euthyroid phenotype but more recently were found to have reduced motor coordination skills when compared with wild type mice.50 Mice homozygous for the Titf-1 gene knockout were born dead, lacking lung parenchyma, with absent thyroid and entire pituitary glands, and with extensive defects in the ventral forebrain.
Not unexpectedly, TTF-1 is expressed in the lungs and ventral forebrain, in addition to the thyroid gland.51 It is known to regulate the transcription of TG and TPO genes, the TSHR gene in thyroid follicular cells,52,53 and the surfactant protein B (SPB) gene in epithelial lung cells.54 A role for TTF-1 in human respiratory development has been borne out by reports of heterozygous de novo TITF-1 deletions (14q13–21 and 14q12–13.3, encompassing the TITF-1 locus) and a mutation (at the 3′ splice consensus of intron 2) inherited in an autosomal dominant manner being associated with compensated congenital hypothyroidism and unexplained respiratory distress in term babies in the neonatal period who had normal bronchial morphology.55–57 The probands had normal sized eutopic thyroid glands on radionuclide and ultrasound scanning, and asymmetrical 99mTc uptake was noted in one report.57 The other prominent recurrent features associated with either de novo or dominantly inherited TITF-1 mutations (including one interstitial deletion in chromosome 14q) are neurological and include hypotonia, persistent ataxia and dysarthria, microcephaly, choreoathetosis, and global developmental delay, suggesting a role for TTF-1 in human brain development.57–59 Major feeding difficulties were also noted. It is thought that the unfavourable neurological outcome was most probably related to TTF-1 deficiency in the thyroid and brain rather than to inadequately corrected congenital hypothyroidism or inadequate thyroxine replacement in an affected mother during pregnancy. Further data published recently suggest that TITF-1 mutations, leading to haploinsufficiency, are also associated with isolated benign hereditary chorea, an autosomal dominant movement disorder.50
Pax-8 is a transcription factor that is one of the nine members of the mammalian paired homeodomain family which recognise DNA through the conserved paired domain and are homologous to the Drosophila segmentation genes.60PAX genes perform key functions in mammalian embryonic development, during which they show highly restricted temporal and spatial expression patterns.61 Pax-8 has a DNA binding domain at the amino terminal end, a carboxy terminal transcriptional activation domain, and a central homeodomain.62 The PAX-8 gene maps to human chromosome 2q12-q14 and consists of 11 exons.63 Studies have shown that Pax-8 plays a fundamental role not only in the initiation of thyroid cell differentiation but also in the maintenance of the differentiated state, and that it is essential for thyroid cell proliferation.64 Once again, mouse models have been instrumental in demonstrating the critical role of Pax-8 in thyroid organogenesis and thyroid cell differentiation. Pax-8 null mice have hypoplastic thyroid glands with absent median anlage derivatives (that is, follicular cells), whereas lateral anlagen derivatives (parafollicular calcitonin producing C cells) are present.65
The survival of homozygous mutant mice depends on thyroxine replacement therapy, whereas heterozygous mice do not display an obvious thyroid phenotype. In contrast, heterozygous human PAX-8 mutations have been described in patients with congenital hypothyroidism of varying severity in six families: four familial cases where congenital hypothyroidism appears to be inherited in an autosomal dominant manner and two sporadic cases.66–69 The thyroid glands in these patients are hypoplastic and sometimes ectopic in location. In one family, the thyroid gland was of normal size at birth but failed to develop normally postnatally, becoming hypoplastic.69 In this family, the probands also showed decreased iodide trapping, with a positive perchlorate discharge test in one, initially suggestive of dyhormonogenesis as a basis for congenital hypothyroidism.
TPO is known to be particularly dependent on Pax-8 for efficient transcription, and therefore reduced TPO expression secondary to impaired Pax-8 function may be an explanation for the partial organification defect.64 Renal hemiagenesis was also reported in two affected cases, one being associated with hypercalciuria.59,69 When present, renal agenesis was ipsilateral to thyroid hemiagenesis.59 Thus congenital hypothyroidism caused by PAX-8 mutations can occur as non-syndromic or syndromic congenital hypothyroidism. Outside the thyroid gland, Pax-8 is also expressed in the kidney, where it is known to activate the Wilms’ tumour gene (WT1) promoter, and in the developing brain.70 Marked phenotypic variability has been found within affected families, suggesting varied penetrance and expressivity of PAX-8 gene defects.
All mutations to date have been located in the conserved paired domain of PAX-8, and the mutant proteins have been shown to have markedly reduced DNA binding with subsequent loss of transcriptional activation function. At the structural level, these mutations are thought to disrupt the pronounced gain of α helical content following interaction of Pax-8 with DNA—that is, impairment of the unstructured to structured transition that occurs during DNA recognition (loss of “induced fit”).68 Pax-8 activates transcription of TPO, TG, and NIS, and acts synergistically with TTF-1 to activate the promoter of human TG gene.70–72 Dominant negative properties of the mutant allele or haploinsufficiency are not considered likely underlying mechanisms for pathogenicity of Pax-8 mutations. The exact mechanism is still unclear but one possibility is that the human PAX-8 locus is imprinted, with selective expression of the mutant allele leading to disease.
The stimulatory G protein α subunit gene (GNAS1) is located on chromosome 20q13 and contains over 13 exons that encode Gsα, the α subunit of the heterotrimeric stimulatory G protein which has intrinsic GTPase activity.73 G proteins mediate signal transduction across cell membranes, coupling extracellular receptors—including those binding TSH, TRH, parathyroid hormone (PTH), and luteinising hormone—to intracellular effector proteins such as ion channels and the adenylyl cyclase and phospholipase C second messenger systems.74 Heterozygous inactivating mutations in GNAS1 result in Albright hereditary osteodystrophy. This is an autosomal dominant disorder characterised by recognisable dysmorphic features including short stature, shortened fourth and fifth metacarpals and metatarsals, obesity, subcutaneous ossification, intracranial calcification, and variable mental retardation. Presumably owing to imprinting mechanisms there is an apparent parent of origin effect whereby maternal transmission usually leads to a pseudohypoparathyroidism type Ia (PHPIa) phenotype, while mutations of paternal origin result in pseudopseudohypoparathyroidism (PPHP).75 PHPIa consists of end organ resistance to multiple hormones including PTH and TSH, leading to low serum calcium and raised levels of phosphate, PTH, and TSH, whereas PPHP patients have Albright hereditary osteodystrophy with a normal biochemical profile. The degree of TSH resistance tends to be mild, and overt clinical hypothyroidism is not always present. The TSH response to TRH is exaggerated and the degree of TSH resistance may increase with age.76
DYSHORMONOGENESIS AND CONGENITAL HYPOTHYROIDISM
The end product of a normally developed hypothalamic-pituitary-thyroid axis is the production of thyroid hormones. Iodide is actively transported and concentrated in the thyroid gland by the sodium iodide symporter present in the basolateral membrane of thyroid follicular cells (fig 3). Subsequently it is oxidised by hydrogen peroxide and bound to tyrosine residues in thyroglobulin to form iodotyrosine (iodide organification). Some of these hormonally inert iodotyrosine residues (monoiodotyrosine and diiodotyrosine) couple to form the hormonally active iodothyronines, T4 and T3. Thyroid peroxidase (TPO) catalyses the oxidation, organification, and coupling reactions. Defects in any of these steps lead to dyshormonogenesis which typically manifests as congenital hypothyroidism and goitre,77 and the responsible gene at each step has been cloned. Except in rare cases, all mutations in these genes appear to be inherited in an autosomal recessive fashion.

Schematic diagram of a follicular cell, illustrating the steps involved in thyroid hormone synthesis. TSH receptor (TSHR) bound to TSH stimulates iodide transport into the thyroid gland by the sodium iodide symporter (NIS). Subsequently, iodide is oxidised by hydrogen peroxide, generated by the recently discovered NADPH oxidase system (ThOX) and bound to tyrosine residues in thyroglobulin (TG) to form iodotyrosine (iodide organification). Some of these hormonally inactive iodotyrosine residues (monoiodotyrosine and diiodotyrosine) couple to form the hormonally active iodothyronines, T4 and T3. Thyroid peroxidase (TPO) catalyses the oxidation, organification, and coupling reactions. The exact function of pendrin, a chloride-iodide transporter, in thyroid hormone synthesis is as yet unknown but it is thought to transport iodide into the colloid from the thyrocyte. Defects in any of these steps lead to dyshormonogenesis, which manifests clinically as congenital hypothyroidism with goitre.
TPO, the enzyme responsible for iodide oxidation, organification, and iodotyrosine coupling, is a 933 amino acid, membrane bound, glycated, haem containing protein, located on the apical membranes of the thyroid follicular cell.77 The human TPO gene is located on chromosome 2p25 and covers approximately 150 kb of DNA.78 The most prevalent cause of dyshormonogenesis is TPO deficiency.79 Defects in the TPO gene have been reported to cause congenital hypothyroidism by a total iodide organification defect.80 Increasing numbers of mutations—including maternal isodisomy for chromosome 2p—have been reported, occurring throughout the 17 exons of the thyroid peroxidase gene, each resulting in an inactive TPO protein.80–91 There is one report of metastatic thyroid carcinoma arising within a congenital goitre associated with a mutation in the TPO gene.85
Thyroglobulin is a homodimer with subunits of 330 000 Da, which is synthesised exclusively in the thyroid gland. The human gene is greater than 300 000 bp in size and is located on chromosome 8q24.92 The coding sequence is divided into 42 exons, each of about 200 bp in size, except that exon 9 and 10 contain 1101 p and 588 bp, respectively.77 Thyroglobulin defects arising from mutations in the gene are associated with moderate to severe congenital hypothyroidism, usually with low serum thyroglobulin concentrations.93 Affected individuals often have abnormal iodoproteins in their serum, especially iodinated albumin, and they excrete iodopeptides of low molecular weight in the urine.77 There is ineffective formation of T4 and T3 resulting from a coupling defect. Owing to its size (coding sequence of 8244 bp), sequencing of the TG gene has been difficult. However, increasing numbers of mutations are being reported.94,95 Thyroglobulin is normally exported through the secretory pathway into the lumen of thyroid follicles to undergo iodination. Probands with deficient thyroglobulin resulting from an altered TG coding sequence have shown defective trafficking of thyroglobulin from the endoplasmic reticulum to Golgi, leading to an endoplasmic reticulum storage disease (ERSD).96 A particular feature of some TG mutations involving cysteine residues in the protein is disruption of the three dimensional structure of the molecule, causing it to be retained in the endoplasmic reticulum as aggregates.96 The cog/cog mouse with a point mutation in the tg gene, in a region which is strictly conserved in the thyroglobulin proteins from all known species, is an example of an ERSD and a model for severe congenital hypothyroidism associated with colloid deficient goitre and abnormal growth and central nervous system development.97 These mice have aberrant folding, dimerisation, and export of thyroglobulin, leading to an abnormally distended endoplasmic reticulum, comparable to that observed in thyroid biopsies from children with congenital goitre from defective thyroglobulin synthesis.98,99
NIS is an integral membrane protein of approximately 65 kDa with 12 potential transmembrane domains,100 with both carboxy and amino termini located inside the cell. Its expression has not only been detected in normal and neoplastic thyroid but also in salivary glands, gastric mucosa, breast, colon, ovaries (lower species), placenta, skin, and choroid plexus.101 The human gene (NIS) has been mapped to chromosome 19p, and the coding region contains 15 exons encoding a protein of 643 amino acids.101 Before the cloning of NIS, a clinical diagnosis of hereditary iodide transport defect had been made for several decades on the basis of goitrous hypothyroidism and absent thyroidal radioiodine uptake.102 The first demonstration of a loss of function mutation in the NIS gene was reported in 1997, following which several mutations inherited in an autosomal recessive manner have been described.103 The hypothyroidism is of variable severity (ranging from fully compensated to severe) and goitre is not always present. Individuals with a higher dietary iodine intake are less likely to have severe hypothyroidism than those with iodine deficient diets.104 This condition is therefore preferably treated with iodine supplementation rather than thyroid hormone replacement. People who are heterozygous for a mutation are euthyroid.103 The loss of function associated with some of these mutations (Q267E and S515X) was accounted for by failure of membrane targeting by the transporter.105
Pendred syndrome is an autosomal recessive disorder characterised by sensorineural deafness and goitre that was first described by Vaughn Pendred in 1896. The incidence of the disease is estimated to be 7.5 to 10 in 100 000, and it is thought to account for as many as 10% of cases of hereditary deafness, making it the most common cause of syndromic deafness.106 Deafness is classically associated with a Mondini cochlear defect, consisting of a reduced number of turns in the cochlea, together with an enlarged vestibular aqueduct; these features are typically present at birth.107,108 Thyroid disease usually presents as a multinodular or diffuse goitre of varying size in most affected individuals, and is typically not evident until the second decade of life. Despite the goitre, individuals are likely to be euthyroid and only rarely present with congenital hypothyroidism.109 TSH levels, however, are often in the upper end of the normal range, and hypothyroidism of variable severity may eventually develop.106 Intrafamilial phenotypic variation has also been noted.110 Affected subjects usually have a positive perchlorate discharge test, with more than 15%—but not complete—release of radiolabelled iodine following perchlorate administration, indicating a mild thyroid organification defect.106 A normal discharge test, on the other hand, does not exclude the diagnosis.110 The PDS gene is on chromosome 7q, contains 21 exons, and is found to be expressed in the cochlea as well as in the thyroid.111 It encodes pendrin, a 780 amino acid protein (molecular weight 86 kDa) with 11 transmembrane domains which functions as a chloride-iodide transporter.112 In contrast to the basolateral cell surface expression of NIS, pendrin has been localised to the apical membrane of a subset of thyroid follicles.112 It has been hypothesised that pendrin transports iodide across the apical membrane of the thyrocyte into the colloid space and that this process is disrupted in Pendred syndrome, such that iodide is taken up normally by the thyrocyte but is not efficiently bound to thyroglobulin in the colloid.113
NADPH oxidases—encoded by two recently cloned genes, THOX1 and THOX2, located at the apical membrane of thyrocytes—are involved in H2O2 generation in the thyroid.114 Both genes are highly expressed in thyrocytes.114 Thyroid peroxidase (TPO) has no biological activity in the absence of H2O2, which is likely to be the limiting factor for thyroglobulin iodination when iodide supply is normal.115 The two human THOX genes are arranged in a head to head configuration and are separated by a 16 kb long region.116 They span 75 kb and are composed of 35 and 34 exons, respectively. How THOX participates in Ca2+/NADPH dependent H2O2 generation in thyroid tissue is still unknown. Defects in the human THOX2 have recently been reported, with heterozygous truncation mutations being associated with mild transient congenital hypothyroidism and a partial iodide organification defect, whereas homozygosity for such defects was associated with severe congenital hypothyroidism and a complete iodide organification defect.117 Goitre was not detected in these patients.
IODOTHYRONINE TRANSPORTER DEFECTS AND SYNDROMIC CONGENITAL HYPOTHYROIDISM
TSH secreted by thyrotrophs in the anterior pituitary gland stimulates the gland to synthesise and secrete thyroid hormones, principally thyroxine (T4). T4 is essentially a prohormone as it is converted to the biologically more active hormone triiodothyronine (T3) by 5′-monodeiodination in all tissues. In a negative feedback loop, T3 suppresses TSH secretion. Thyroid hormone exerts its effects by acting on nuclear receptors, necessitating transmembrane passage of the hormone. Several classes of membrane thyroid hormone transporters have been identified recently, including MCT8, which was mapped to Xq13.2 and contains five exons.118 There are recent reports of mutations in this gene found in male individuals in four families with abnormal thyroid function tests consisting of low FT4 and raised levels of FT3 and TSH, which were not found at the time of neonatal screening, but detected within the first two years of life.119 Stigmata of congenital hypothyroidism were not present at birth. In addition, neurological abnormalities from early infancy were described, consisting of central hypotonia, peripheral hypertonia, dystonia, rotary nystagmus, dysconjugate eye movements, feeding difficulties, vomiting, recurrent aspiration, irritability, and subsequent spastic quadriplegia resulting in severe delay in motor and mental development with absent speech reported in one proband. Brain MRI and electroencephalography were normal. Thyroid imaging has not been described. Female relatives harbouring mutations in MCT8 displayed milder thyroid phenotypes without the neurological features. Expression of this gene has been found in all tissues examined, including brain, thyroid, pituitary, and placenta.119 This is the first example of X linked congenital hypothyroidism resulting from a defect in target tissue rather than in the pituitary-thyroid biosynthetic pathway.
INVESTIGATION AND MANAGEMENT OF CONGENITAL HYPOTHYROIDISM
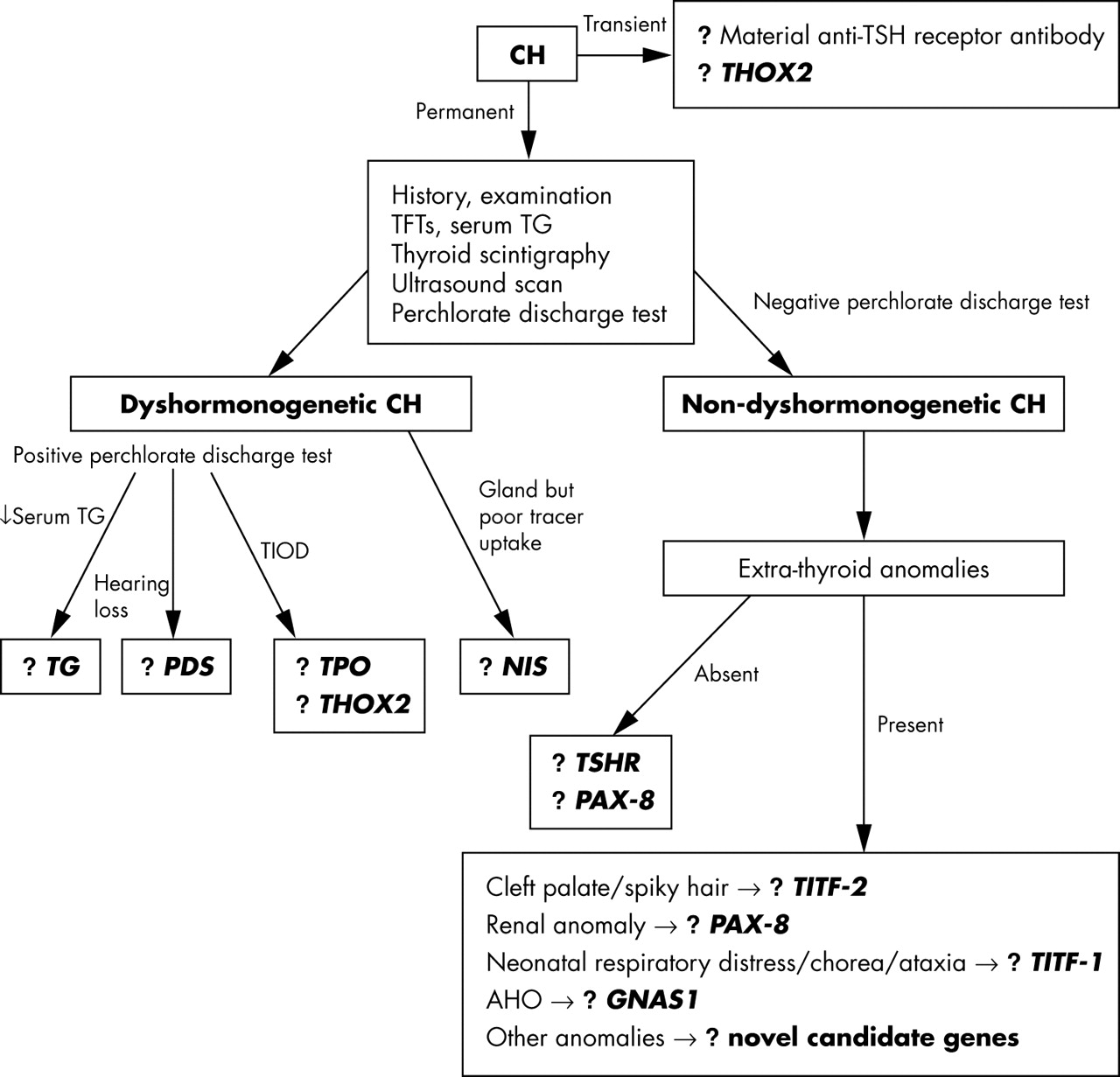
In those cases of congenital hypothyroidism where an underlying genetic defect is suspected—on the basis of family history, evidence of dyshormonogenesis, or the presence of other congenital anomalies—to suggest a syndromic form of the disorder, further investigation can prove fruitful. While the outcome of such genetic analysis may not necessarily affect the management of the index patient, the results can aid genetic counselling regarding recurrence risk within the family and may suggest the best treatment option—for example, congenital hypothyroidism from NIS defects is more amenable to treatment with iodide supplementation than with thyroxine. We suggest below some investigations to aid in the search for an underlying genetic cause in a small number of cases with congenital hypothyroidism. They are not intended to be a comprehensive list of recommended investigations for physicians managing routinely diagnosed cases.
As congenital hypothyroidism is largely a sporadic disorder, a family history is not present in most cases. However, a detailed history should be taken, including that of parental consanguinity, other affected family members, and a history of any extrathyroidal congenital anomalies (for example, cleft palate, renal anomalies, neonatal respiratory distress, and movement disorders). A neonate born with severe congenital hypothyroidism may have the associated features of prolonged jaundice, macroglossia, hoarse cry, and umbilical hernia on examination. The neck should be examined for a goitre and its presence would suggest dyshormonogenesis as the basis of congenital hypothyroidism. Rarely, the goitre can be detected in utero by ultrasound scanning, and such cases can be treated antenatally by intra-amniotic injections of thyroxine or triiodothyronine, which can successfully shrink the gland.120 If a significantly enlarged goitre escapes antenatal detection, there could be acute problems postnatally if the upper airway is compromised, and the expert opinion of a paediatric ENT specialist is vital in such cases. The presence of any other congenital malformations should also be sought on examination. Delayed maturation of bone on x ray is of limited value in infancy as the ossification centres are composed mainly of cartilage; it is of greater value after two years in severe forms of congenital hypothyroidism. The initial crucial steps in investigating congenital hypothyroidism are to establish the severity of the hormone defect, to investigate for the possibility of dyshormonogenesis, and determine the morphology and position of the thyroid gland (fig 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A proposed algorithm for investigating the genetic basis of congenital hypothyroidism. AHO, Albright hereditary osteodystrophy; CH, congenital hypothyroidism; GNAS, stimulatory G protein α subunit gene; NIS, sodium-iodide symporter gene; PAX-8, human Pax-8 gene; PDS, Pendred syndrome gene; TFTs, thyroid function tests; TG, thyroglobulin gene; THOX2, thyroid oxidase 2; TIOD, total iodide organification defect; TITF-1, human TTF-1 gene; TITF-2, human TTF-2 gene; TPO, thyroid peroxidase gene; TSHR, TSH receptor gene; USS, ultrasound scan.
Biochemical investigations
The first of these steps is established by obtaining the original diagnostic thyroid function tests from the neonatal blood spot screening card and from the first venous serum measurements, before the onset of L-thyroxine treatment. In up to 10% of cases, the congenital hypothyroidism is transient and self limiting, lasting up to a few months.121 Some of these cases can be accounted for by the presence of anti-TSHR antibodies in the mother, with transplacental passage to the fetus. When detected, the function of these TSHR autoantibodies should be measured, as there are reports of hyperthyroid mothers with Graves’ disease giving birth to hypothyroid infants.122
In other infants with transient congenital hypothyroidism and an abnormal perchlorate discharge test suggestive of a defect in organification within the thyroid, the possibility of heterozygous mutations in THOX2 should be considered. The measurement of serum thyroglobulin is also useful as all patients with organification disorders, and cases with severe TSHR defects, have raised levels when their circulating TSH is increased, except in cases with thyroglobulin synthesis defects where the thyroglobulin level is usually low. Thyroglobulin measurements can provide useful additional information about the thyroid gland—for example, if serum thyroglobulin is measurable where there is apparent athyreosis on thyroid imaging, this suggests that radiologically undetectable hypoplastic thyroid tissue is likely to be present.32
It is also worth checking thyroid function (circulating TSH, FT4, and FT3) and thyroglobulin levels not only in affected family members but also in all first degree relatives of probands, as variable penetrance associated with autosomal inheritance might lead to the identification of a mildly affected relative not detected previously. Furthermore, with TSHR mutations, heterozygotes may also display a mild phenotype. Underlying autoimmune thyroid disease, causing raised TSH with low or normal thyroid hormones, can be excluded by the absence of thyroid autoantibodies. A useful test to determine whether the thyroid gland is resistant to TSH involves the administration of TRH, with measurement of both the pituitary TSH response and the subsequent rise in circulating T3 following this increase in TSH.
Defective iodide trapping by the thyroid gland caused by NIS mutations can be tested for by measuring the saliva to plasma (S/P) ratio of iodide one hour after oral administration of a small dose of radiolabelled iodide (between 25 and 50 μCi of either 125I or 131I labelled NaI). The normal S/P ratio is 25 to 140.123 Thyroxine treatment need not be stopped or reduced before this test, as it does not affect salivary gland iodide trapping.
Radiological investigations
Thyroid scintigraphy, using either technetium (Tc) or radioactive iodine, is usually the first line imaging in many centres and provides information about the size and location of the gland, but it can be misleading. For example, it can indicate no uptake of isotope, suggesting apparent athyreosis, although a thyroid gland is present. Ultrasound scanning of the neck by an experienced radiologist can provide further valuable information, with assessment of whether thyroid gland size is appropriate for age and sex, and its location along the embryological line of development between the base of the tongue and thorax. Ultrasound scanning in conjunction with isotope scanning can provide further useful information. For example, if there is discordance between thyroid isotope scanning and ultrasound scanning, with the former showing no tracer uptake yet the latter showing the presence of a normal or even an enlarged thyroid gland, there is strong evidence for the involvement of NIS causing congenital hypothyroidism. Based on history and initial investigations, further imaging can be undertaken, such as a renal ultrasound scan in a family with autosomal dominant congenital hypothyroidism associated with thyroid gland hypoplasia (?Pax-8 defect), or MRI of the inner ear in cases of congenital hypothyroidism with goitre and hearing loss. The proportion of radioiodine discharged from the thyroid following the administration of potassium perchlorate is an index of the severity of organification defects. Discharge of about 20–45% indicates a partial and mild defect, but values greater than 60% and 90% are typical of severe and complete defects, respectively.80
Treatment
Once the diagnosis of congenital hypothyroidism is made on neonatal screening, appropriate thyroxine replacement therapy is initiated by a paediatrician. Studies have shown that several variables influence eventual IQ in children with congenital hypothyroidism. These include the severity of congenital hypothyroidism (as determined by thyroid function tests and delayed skeletal maturation at birth), the dose of thyroxine treatment, the timing of treatment onset, and serum free thyroxine concentrations during the first year.124–127 Despite the establishment of neonatal screening programmes, clinicians and families should be aware of reports suggesting that about 10% of early treated infants with severe hypothyroidism are likely to require special education.128 Therefore clinically significant intellectual impairment should be actively screened for and treated when detected in these children.
CONCLUSIONS
Congenital hypothyroidism represents a common neonatal problem that is easily detected and treated, and hence the success of the nationwide neonatal screening programmes. While the great majority of cases are considered sporadic, there have been recent advances in elucidating some of the molecular mechanisms behind certain forms of this common congenital metabolic disorder. Organification defects leading to goitrous congenital hypothyroidism are now well known to have an autosomal recessive genetic basis. More recently, there is growing evidence for the role of germline gene defects causing congenital hypothyroidism associated with thyroid dysgenesis. These include a role for the TSHR gene in non-syndromic congenital hypothyroidism, with evidence for its involvement in recessive disease, and possibly also heterozygous mutations in this gene in congenital or even acquired non-autoimmune hypothyroidism. Downstream of TSHR, defects in Gsα result in TSH resistance in Albright’s hereditary osteodystrophy. Defects in the transcription factors TITF-2 (cleft palate, spiky hair), TITF-1 (neonatal respiratory distress, involuntary movement), and Pax-8 (renal hemiagenesis) provide the basis for multisystem involvement in syndromic forms of congenital hypothyroidism. Finally, there is early evidence emerging for a third group of congenital hypothyroid conditions associated with defects in target tissue iodothyronine transporters, with devastating neurological features.
Novel and as yet unidentified target genes regulated by these transcription factors may also prove to be important candidate genes in other forms of congenital hypothyroidism. A role for the transcription factor Hoxa3 (human locus 7p15.1), a member of the Hox family of homeobox genes involved in regulating the development of pharyngeal glandular organs, has hitherto only been shown in murine embryonic thyroid development, with null mice having thyroid hypoplasia resulting from defects in both follicular and parafollicular cell development.129 Interestingly, these mice also have other abnormalities, including thymic and parathyroid aplasia, anomalies of the heart and great vessels, and malformations of the throat and jaw, which are remarkably similar to those observed in 22q11 deletion (velocardiofacial) syndrome in humans.130 It is also known that the amount of thyroid tissue in these patients is variably reduced, as in the Hoxa3 mutant mice,131 and it has therefore been suggested that patients with 22q11 deletion syndrome may be at increased risk of thyroid dysfunction. Hoxa5(−/−) mutant mice have been reported to have hypothyroidism associated with transient growth retardation, delayed eye opening, and ear elevation.132 Thyroid gland development begins normally, but follicle formation and thyroglobulin processing are abnormal in late gestation. The expression of several molecular markers essential for thyroid gland formation and function—namely TTF-1, Pax8, and TTF-2—is affected in the developing thyroid gland, with the consequence of altered expression of thyroid effector genes, including the thyroglobulin and TPO genes. Murine Nkx-2.5, a member of the homeobox gene superfamily that is related to the TITF1 (NKX-2.1), is expressed early during embryogenesis of both thyroid and myocardium,133 and accordingly represents a strong candidate in those 4% of cases of congenital hypothyroidism that are associated with cardiac defects.134
With regard to identification of additional novel candidate genes, future directions include genome-wide linkage analyses in large families with multiple probands or parental consanguinity, identification of congenital hypothyroidism phenotypes in transgenic or randomly mutagenised mouse models, and linkage analysis and positional cloning in well characterised contiguous gene syndromes with congenital hypothyroidism as a prominent feature (for example, William syndrome (7q11 deletion) where congenital hypothyroidism occurs in 25% of affected individuals). Known candidate genes will have to be carefully ruled out in investigating for this genetically heterogeneous condition where phenocopies pose difficulties—for example, in the absence of a family history it may be difficult to distinguish between TSHR mutations and Pax-8 defects affecting only the thyroid.
Acknowledgments
SMP is a recipient of a Wellcome Trust research training fellowship for medical and dental graduates and the Raymond and Beverly Sackler studentship from the University of Cambridge School of Clinical Medicine.
REFERENCES
Footnotes
-
Competing interests: none declared