Article Text

Abstract
Background: The haplotype H1 of the tau gene, MAPT, is highly associated with progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD).
Objective: To investigate the pathogenic basis of this association.
Methods: Detailed linkage disequilibrium and common haplotype structure of MAPT were examined in 27 CEPH trios using validated HapMap genotype data for 24 single nucleotide polymorphisms (SNPs) spanning MAPT.
Results: Multiple variants of the H1 haplotype were resolved, reflecting a far greater diversity of MAPT than can be explained by the H1 and H2 clades alone. Based on this, six haplotype tagging SNPs (htSNPs) that capture 95% of the common haplotype diversity were used to genotype well characterised PSP and CBD case–control cohorts. In addition to strong association with PSP and CBD of individual SNPs, two common haplotypes derived from these htSNPs were identified that are highly associated with PSP: the sole H2 derived haplotype was underrepresented and one of the common H1 derived haplotypes was highly associated, with a similar trend observed in CBD. There were powerful and highly significant associations with PSP and CBD of haplotypes formed by three H1 specific SNPs. This made it possible to define a candidate region of at least ∼56 kb, spanning sequences from upstream of MAPT exon 1 to intron 9. On the H1 haplotype background, these could harbour the pathogenic variants.
Conclusions: The findings support the pathological evidence that underlying variations in MAPT could contribute to disease pathogenesis by subtle effects on gene expression and/or splicing. They also form the basis for the investigation of the possible genetic role of MAPT in Parkinson’s disease and other tauopathies, including Alzheimer’s disease.
- CBD, corticobasal degeneration
- CEPH, Centre d’Etude du Polymorphisme Humain
- EM, expectation maximisation
- FTD, frontotemporal dementia
- FTDP-17T, frontotemporal dementia with parkinsonism with tau pathology linked to chromosome 17
- htSNP, haplotype tagging single nucleotide polymorphism
- LD, linkage disequilibrium
- LRT, likelihood ratio test
- MAPT, microtubule associated protein, tau
- MIM, mendelian inheritance in man
- PSP, progressive supranuclear palsy
- RFLP, restriction fragment length polymorphism
- SNP, single nucleotide polymorphism
- tau
- tauopathy
- parkinsonism
- haplotype
- progressive supranuclear palsy
Statistics from Altmetric.com
- CBD, corticobasal degeneration
- CEPH, Centre d’Etude du Polymorphisme Humain
- EM, expectation maximisation
- FTD, frontotemporal dementia
- FTDP-17T, frontotemporal dementia with parkinsonism with tau pathology linked to chromosome 17
- htSNP, haplotype tagging single nucleotide polymorphism
- LD, linkage disequilibrium
- LRT, likelihood ratio test
- MAPT, microtubule associated protein, tau
- MIM, mendelian inheritance in man
- PSP, progressive supranuclear palsy
- RFLP, restriction fragment length polymorphism
- SNP, single nucleotide polymorphism
The tauopathies are a group of neurodegenerative disorders that are characterised pathologically by fibrillar aggregates of the microtubule associated protein, tau. These disorders include Alzheimer’s disease, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease, and frontotemporal dementia with parkinsonism with tau pathology linked to chromosome 17 (FTDP-17T), with a clinical spectrum ranging from dementia to parkinsonian phenotypes.1 The identification of missense and splice site mutations in the tau gene, MAPT (MIM 157140), causing FTDP-17T (MIM 600274) affirmed a central role for tau dysfunction in some neurodegenerative diseases.2,3 Although the other related tauopathies—including Alzheimer’s disease, PSP, and CBD—are defined by fibrillar tau pathology, MAPT is not mutated in these diseases.
PSP (MIM 601104; Steele–Richardson–Olszewski syndrome)4 is usually a sporadic disorder of late adult life. It is the second most common form of degenerative parkinsonism and is characterised clinically by an akinetic-rigid syndrome, supranuclear gaze palsy, pseudobulbar signs, and cognitive decline of frontal lobe type.5–7 CBD is an atypical parkinsonian condition occurring much less commonly than PSP and it classically presents with unilateral cortical sensory loss, alien hand, jerky dystonia, rigidity, bradykinesia, and dementia. PSP is sporadic, with no familial history or MAPT mutations in the large majority of cases. However, robust genetic association of PSP with MAPT and reports of the rare families with more than one affected member8,9 indicated that genetic factors could play a role. Conrad and colleagues were the first of many groups to show that variation at the MAPT locus could be an important genetic influence in sporadic PSP by demonstrating allelic association with PSP of a dinucleotide polymorphism in MAPT intron 9.10 The overrepresentation of the commoner allele (a0) in PSP and also later in CBD was then confirmed by other groups.11,12 This suggests either that this polymorphism itself could contribute to increased risk or that it is in linkage disequilibrium (LD) with the actual causative variant. Although some MAPT mutations in FTDP-17T cause a clinical picture closely resembling PSP,13–15 no pathogenic variations of MAPT have yet been identified in clinically and pathologically diagnosed sporadic and familial PSP.16
The allelic association of MAPT with PSP and CBD was subsequently extended to a series of polymorphisms extending over the entire MAPT coding region spanning nearly 62 kilobases (kb).17 In approximately 200 unrelated white subjects, these polymorphisms were in complete LD, forming two extended haplotypes, H1 and H2.17 The study suggested that the establishment of these two haplotypes was an ancient event and that either recombination was suppressed in this region, or recombinants were selected against. It also showed that the more common haplotype, H1, with which the a0 allele segregated, was significantly overrepresented in PSP.17 Follow up studies18,19 extended the MAPT haplotype a further 68 kb to the promoter region of MAPT where three SNPs, highly associated with PSP, were in complete LD with the rest of the MAPT haplotype.19 We have further extended the MAPT haplotype to cover a maximal region of ∼2 million bases (Mb) which is in near complete LD,20 and using high density HapMap genotype data for LD analysis we subsequently revised the size of the region to 1.8 Mb (unpublished work). This region associated with PSP includes several other genes in addition to MAPT, including Saitohin21,22 (situated within intron 9 of MAPT), NSF (N-ethylmaleimide sensitive factor), IMP5 (a presenilin homologue),23CRHR1 (corticotrophin releasing hormone receptor), and LOC284058, an unknown gene just adjacent to MAPT.
Identifying the functional basis of the H1 haplotype association will be important in providing an insight into the aetiopathogenesis of PSP and CBD. Although all the genes within this multigene haplotype block are associated with PSP and CBD, the hallmark tau pathology of these disorders strongly implicates MAPT itself. The aim of our study was therefore to analyse exhaustively the MAPT haplotype association with PSP and CBD in order to identify non-coding variants that could affect tau gene expression, splicing, or processing, leading to tau pathology and selective neuronal loss. More controversially, recent work shown weak association of the H1 haplotype with sporadic Parkinson’s disease24 and association with Norwegian Parkinson’s disease cases of a haplotype within the extended H1 clade, spanning the 5′ half of MAPT.25 This is surprising as Parkinson’s disease is traditionally not associated with tau dysfunction or pathology.
In this work, we employed a systematic framework of genetic analyses to investigate the common haplotype structure of MAPT in order to refine the association of the MAPT haplotype with PSP and CBD. By using the validated high density genotype data available from the International HapMap Project (www.hapmap.org) we analysed the MAPT gene in 27 defined CEPH (Centre d’Etude du Polymorphisme Humain) trios (father, mother, and offspring). We analysed LD and haplotype structure with 24 SNPs in relation to the H1 and H2 haplotypes, as defined by the MAPT biallelic intron 9 deletion-insertion (del-In9),17 using the software suite TagIT (www.popgen.biol.ucl.ac.uk/software.html), which contains routines specifically tailored for the inference of haplotypes from the CEPH trio data.26 With this analysis, we identified far greater haplotypic variation of MAPT than can be explained by the description of the extended H1 and H2 haplotypes alone. Based on the data for this common haplotypic diversity of MAPT in the CEPH trios, we identified a set of six haplotype tagging SNPs (htSNPs): five SNPs that represent intra-H1 variation and del-In9.17 The htSNPs function as a minimal set of highly informative single nucleotide polymorphism (SNP) markers that capture 95% of the common haplotype diversity of MAPT.26 We genotyped the MAPT htSNPs in our target populations, namely well characterised PSP case–control cohorts of both British and north American (US) origins and CBD cases of US origin.
METHODS
Analysis of the linkage disequilibrium and haplotype structure
SNP data for the region of the MAPT locus in 27 CEPH trios (Corriell Institute for Medical Research; http://locus.umdnj.edu/nigms/) from the International HapMap project (HapMap) web site (http://www.hapmap.org/) were downloaded for genetic analysis of the MAPT. The raw SNP genotype data were analysed in TagIT, a software package for identifying and evaluating tagging SNPs applied to haplotype data, which also contains routines for inferring haplotypes from trio material and LD analysis (http://popgen.biol.ucl.ac.uk/software).26
We initially removed from the HapMap data any SNPs that had a minor allele frequency of less than 5%. We also checked for any inconsistencies in the data through the parent–offspring relationship in the CEPH trios. We used a resulting set of 24 SNPs and the del-In9 (table 1) which covers the entire MAPT gene from upstream of the promoter to beyond exon 13, to infer haplotypes and their respective frequencies by an expectation–maximisation (EM) algorithm (ε = 1×10−6) specifically for CEPH trio material (EM trio).26 For convenience, we designated the biallelic (+/−) intron 9 deletion-insertion polymorphism (del-In9) as an SNP. In all, 34 haplotypes were resolved from parental chromosomes. The pairwise LD across MAPT for each SNP was then evaluated by both the measures of D′ and the square of the correlation coefficient (r2). Both measures were calculated, first by estimating pairwise haplotype frequencies through EM trio, then by assessing the statistical strength of association through a likelihood ratio test (LRT), by comparing the EM frequencies with haplotype frequencies estimated assuming no LD. Both measures of LD are based upon D, the basic pairwise disequilibrium coefficient, the difference between the probabilities of observing the alleles independently in the population: D = f(A1B1)−f(A1)f(B1).27 A and B refer to two genetic markers and f is their frequency. D′ is obtained from D/Dmax and a value of 0.0 suggests independent assortment, whereas 1.0 means that all copies of the rarer allele occur exclusively with one of the possible alleles at the other marker. The measure of r2 has a more strict interpretation than that of D′; r2 = 1.0 only when the marker loci also have identical allele frequencies. The allele at the one locus can always be predicted by the allele at the second locus. Recent work suggests that r2 is the preferred measure of LD for association based studies.26
The 24 single nucleotide polymorphisms and del-In9 used for the linkage disequilibrium and haplotype structure analysis of MAPT in the CEPH trios
Allelic and genotype frequencies followed by statistical assessment of Hardy–Weinberg equilibrium were made at each locus in the CEPH trios as implemented by TagIT.
From the LD and haplotype structure of MAPT, htSNPs were selected to capture the diversity of known MAPT HapMap SNPs in the CEPH trios. We selected six tagging SNPs (del-In9, SNPs 8, 14, 17, 21, and 25); using TagIT, we then assessed their performance on the CEPH trios. Our tagging approach focused on the coefficient of determination (that is, haplotype r2) in a linear regression, which uses the haplotypes defined by the htSNPs to predict the state of the tagged SNPs.26 The basis of this design is that even when individual haplotypes defined by the htSNPs do not correlate perfectly with tagged SNPs, haplotype combinations might do so, and these combinations are identified by selection of the appropriate coefficients in the linear regression. Haplotype r2 is the coefficient of determination from an analysis of variance of locus i (coding alleles at locus i as “0” or “1”) among the G groups (number of haplotypes, or groups, defined in the dataset in question by the htSNP set): r2[hap]i = 1−R′i/Di, where R′i = 2Σp′ig(1−p′ig)/xg, which can be interpreted as the sum of the within group variances weighted by their frequency.
The PSP cases and control subjects
The unrelated PSP cases (n = 83), from the Queen Square brain bank for neurological disorders, were all white and of western European origin and were all pathologically confirmed. Most of these cases have been used in previous studies.16,19,20,22,28 Pathological confirmation of the diagnosis of PSP was made following standardised criteria.28 The unrelated British control population (n = 169), all white, were taken from brain bank tissue with no clinical evidence of neurodegenerative disease and no abnormal histopathology, from the MRC Building, Newcastle, UK. The samples were age matched, where the average age at death was 73.5 years for the PSP cases (63% male) and 76 years for the controls (51% male). All patients and controls were collected under approved protocols followed by informed consent, and this work was approved by the joint research ethics committee of the Institute of Neurology and the National Hospital for Neurology and Neurosurgery.
The unrelated US control population consisted of individuals (n = 131; 50% male) free of abnormal histopathology and with an average age at death of 79.9 years. The unrelated PSP cases (n = 238; 50% male) were pathologically confirmed by standard criteria and had an average age at death of 75.3 years. The unrelated CBD cases (n = 44; 50% males) were pathologically confirmed following standard criteria and had an average age at death of 71.3 years.
Genotyping
The htSNPs (dbSNP numbers: rs1467967, rs242557, rs3785883, rs2471738, and rs7521, and the del-In9; table 1) were genotyped in the PSP case–control cohorts as follows. The 238 bp MAPTdel-In9 was genotyped as previously described.17 Polymerase chain reaction (PCR) primer pairs (available on request) were designed by the Primer3 program (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) and used to amplify each SNP of interest. PCR reactions were as follows: 10 μl reactions, which contained one unit of DNA polymerase (Qiagen, Crawley, West Sussex), 10×PCR reaction buffer, 5×Q solution (Qiagen), 10 pmol of each oligonucleotide primer pair, and 25 ng of sample template genomic DNA.
Genotyping of the SNPs rs1467967, rs242557, rs3785883, rs2471738, and rs7521 was conducted by Pyrosequencing (Biotage AB, Uppsala, Sweden) (details available on request) or by restriction fragment length polymorphism (RFLP) digest. The following restriction endonucleases cut the PCR product once at the (N) allele: Dra I (A), ApaL I (A), BsaH I (G), BstE II (T), and Pst I (A) (New England Biolabs, Hitchin, Herts, UK). PCR products were incubated overnight with 2 units of the corresponding restriction enzyme at the recommended temperature. Digests were separated on 4% agarose gels and visualised with ethidium bromide staining.
We assessed genotyping accuracy by retyping 20% of all genotypes, whole sets of htSNPs, genotyping by alternative methods and by direct automated DNA sequencing of random samples.
The ancestral allele at each locus was determined by direct sequence comparison of the 24 SNP loci in human and chimpanzee MAPT and in addition by searching for the ancestral allele in NCBI (http://www.ncbi.nlm.nih.gov/).
Statistical analysis
For each htSNP, the allele and genotype distribution in the PSP cases were compared with those in the control group. Statistical assessments for the allele and genotype frequencies and Hardy–Weinberg were made using TagIT. Case–control single locus htSNP allelic and genotypic association was calculated statistically in CLUMP software.29 The p values were derived by standard Pearson’s χ2 tests except in cases where cell counts in the contingency tables were less than 5. When cell counts were less than 5, p values were determined empirically by 100 000 simulations; the program uses a Monte-Carlo approach that performs repeated simulations to generate random tables having the same marginal totals as the one under consideration and counting the number of times that a χ2 value associated with the actual table is achieved by the randomly generated tables. We tested for heterogeneity between the H1H1 homozygote populations versus the whole population using a standard Pearson χ2 test.
Distributions of haplotypes defined by the htSNPs were compared in the PSP cases and controls using WHAP software (http://www.broad.mit.edu/personal/shaun/whap/). This is an SNP haplotype analysis suite that performs a regression based haplotype association test through an LRT, which is a χ2 test with n−1 degrees of freedom to derive the associated p value, where n is the number of haplotypes observed for the data. We used this test to give an initial assessment of haplotype association (an omnibus test) and then carried out individual haplotype tests (haplotype specific tests) of association, again through an LRT (df = 1) and by also obtaining empirical p values by Monte-Carlo methods (20 000 simulations used). To test the effect of the H1 specific htSNPs while controlling for the extended H1/H2 haplotype we imposed a set of equality constraints under the null across the haplotypes identical at the del-In9 and undertook single locus and haplotype analysis as outlined above. We corrected the p values in tables 4 and 5 according to the number of tests performed where appropriate by the Bonferroni correction, the significance of which is discussed throughout the text.
RESULTS
Linkage disequilibrium and haplotype structure of MAPT
For the haplotype analysis of the MAPT gene, we downloaded genotype data for 27 CEPH trios (mother, father, and offspring) of European descent (CEPH Utah collection) for SNPs spanning the MAPT region, from the International HapMap Project web site (www.hapmap.org). The raw SNP data from HapMap were analysed using the software package TagIT (http://popgen.biol.ucl.ac.uk/software). We discarded SNPs that had a minor allele frequency of less than 5%. No inconsistencies in Mendelian inheritance in the parent–offspring relationship were found. We genotyped the del-In9 marker that defines the extended H1 and H2 clades.17 The average density of the markers is one SNP every 6.7 kb. None of the polymorphisms deviated from Hardy–Weinberg equilibrium. See table 1 for details of all SNPs analysed in the CEPH trios.
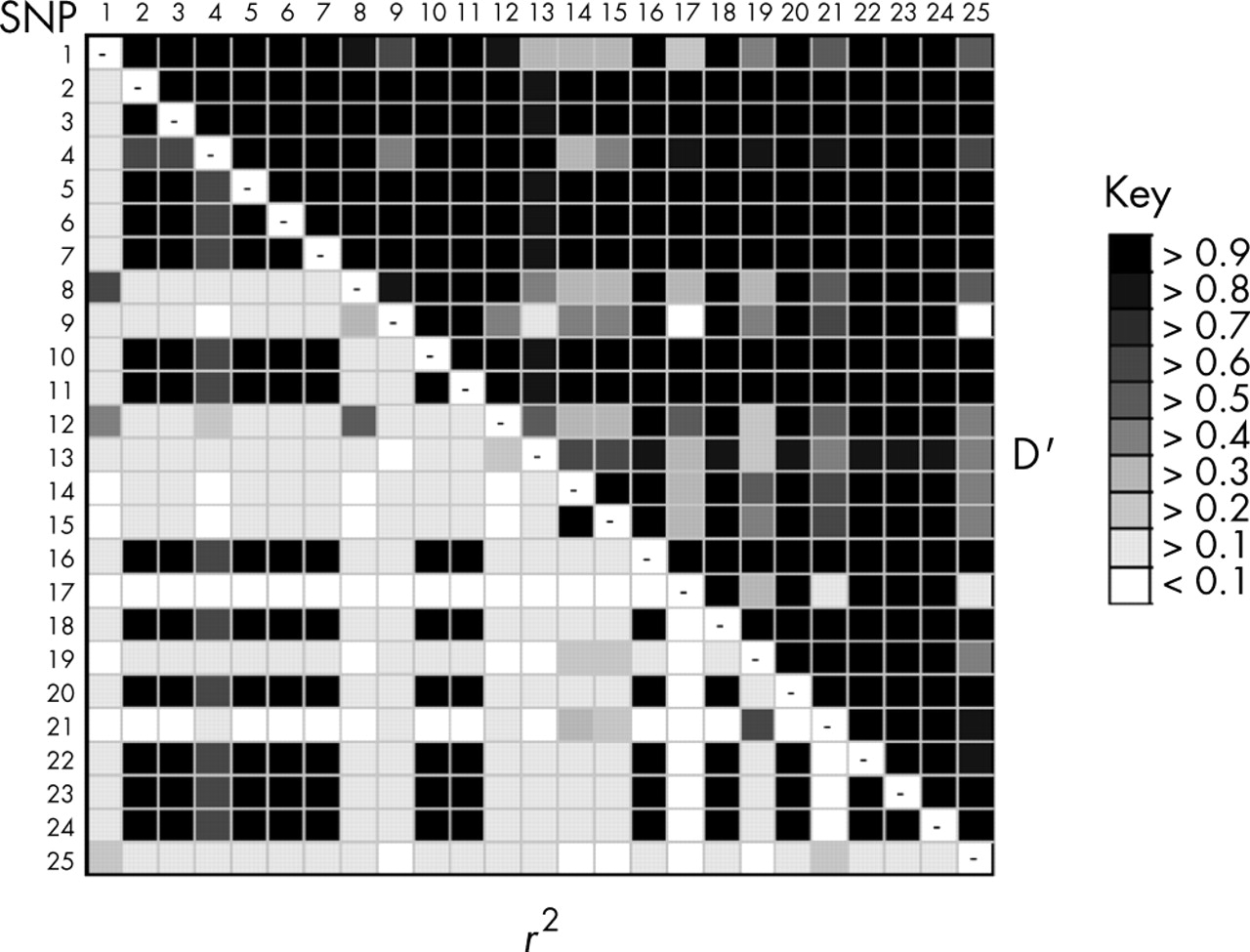
We evaluated pairwise LD across MAPT for all 24 selected SNPs and del-In9 in the 27 CEPH trios both by D prime (D′) and the square of the correlation coefficient (r2), calculated from the expectation-maximisation trio (EM trio) inferred haplotypes. By pairwise LD analysis of the 25 SNPs in CEPH trios, we identified a greater diversity than reflected by the description of the two extended H1 and H2 haplotypes alone (fig 1). The entire MAPT gene is featured by significant LD as is particularly evident by the measure of D′ (fig 1). However, when LD was assessed by the more stringent measure of r2 (which accounts for differences in allele frequencies), it appeared more fragmented, with SNPs that were in high r2 LD with each another, but in moderate to low r2 LD with the extended H1 and H2 haplotype (defined by the del-In9 and other SNP loci), suggesting that they are correlated with either the H1 or H2 haplotypes, but with differing frequency. This supports evidence of variability on the background of these extended haplotypes. In fact, our analyses in the CEPH trios show that these underlying blocks of LD were variable exclusively on the background of the extended H1 haplotype and therefore defined haplotypes within the H1 clade. LD correlation by D′ between many of the described H1 specific SNPs is relatively low (fig 1), suggesting a degree of linkage equilibrium between them; this indicates that, unlike the H1 and H2 haplotypes, there are no constraints to recombination between variants of the extended H1 haplotypes. This pattern of LD across the extended H1 haplotype is essentially similar in the Taiwanese population, in which the extended H2 haplotype is absent (unpublished data).

Linkage disequilibrium (LD) across the MAPT gene. Numerical LD is presented by grey scale, pairwise between each single nucleotide polymorphism (SNP) by both D′ (upper right) and the more stringent measure r2 (bottom left). The darker the shading indicates a greater extent of LD between the SNPs.
We obtained the EM inferred MAPT haplotypes and their respective frequencies by using the EM estimation algorithm specifically tailored to deal with trio data (EM trio) as structured in the CEPH trios.26 We also obtained phased haplotypes (n = 34, representing 42% of the total number of haplotypes in the CEPH trios) by resolving parental chromosomes in the CEPH trios. EM predictions depict a total of 14 different MAPT haplotypes of frequency greater than 1% (table 2). Three of these haplotypes are common, having a frequency greater than 10%, with the remaining 21 haplotypes having frequencies of less than 5%. Only one of the common predicted haplotypes (haplotype A, frequency = 18.1%) is representative of H2 (table 2). The other two common variants (B and C; frequencies = 17.2% and 14.2%, respectively) are based upon the H1 haplotype and differ from one another at multiple SNP loci, as shown in fig 2. A further 11 rare variants of the H1 haplotype (frequency less than 1%) were predicted.
The haplotype structure of the MAPT gene in CEPH-trios based upon the 25 markers in Table 1

Reduced median network of MAPT haplotypes (frequencies that exceed 1% by expectation maximisation, from table 2) in the CEPH trios. Node size is proportional to haplotype frequency. Any two given haplotypes differ by the single nucleotide polymorphism(s) (as numbered from table 1) along the lines that connect them.
It is noteworthy that in addition to the resolved H2 haplotype A, a single resolved haplotype (haplotype G; frequency 2.9% in resolved), based on variation of H2 haplotype A, was resolved which differed from haplotype A by SNP 13 (table 2). However, this haplotype was not predicted by EM trio for output as a significant frequency in the population and represented only ∼5% (estimated by EM prediction) of all H2 haplotypes in the CEPH trios. It is thought that haplotype prediction through EM is a more accurate representation of the relative haplotype frequencies in a population than simply resolving “known” haplotypes because of a far greater utilisation of the data. We also constructed the ancestral (chimpanzee) haplotype based upon the alleles of the 24 SNPs and the del-In9 (table 2). This appears not to resemble any haplotype present in the CEPH trios, though its closest relative (but different by 10 loci) would appear to be that of the extended H2 (CEPH trio haplotype A, from table 2). The other ancestral SNP loci are either consistent with the H1 haplotype family (SNPs 1, 5, 6, 10, 12, 18, and 23), including the presence of the 238 bp insertion sequence (del-In9, or SNP 22 in table 1), or the allele is not observed in Homo sapiens (SNPs 7 and 11).
Selection, performance assessment, and association analysis of MAPT haplotype tagging SNPs
We used an association based criterion (criterion 5 in TagIT, haplotype r2) in order to select the haplotype tagging SNPs (htSNPs).26 Six htSNPs (SNPs 8, 14, 17, 21, 22 (del-In9), and 25; table 1) are sufficient to represent all the HapMap SNPs in the 27 CEPH trios with a high coefficient of determination. Five of these htSNPs are H1 specific—that is, they vary only on the H1 background. In addition the bi-allelic del-In9 marker is used to unambiguously distinguish the extended H1 and H2 haplotypes.17 In CEPH trios26 the performance value for the 6 htSNPs and del-In9 in the CEPH trios was interpreted at an average haplotype r2 value of 0.95 (95%) and a minimum r2, interpreted as the minimum locus value of 0.68. Excluding the del-In9 from the set of htSNPs results in a loss of performance of only of 3%, with performance down to 92% with the five remaining H1 specific htSNPs. This is because a particular allelic combination of these five H1 specific SNPs is representative of the extended H2 haplotype. The performance value of just the del-In9 against the known SNPs in the CEPH trios is just 50%.
We genotyped the MAPT htSNPs in two separate PSP case–control cohorts from the UK and USA and CBD cases from USA. Single locus association results are summarised in table 3. In none of the groups were there any significant deviations from Hardy–Weinberg equilibrium at any of the htSNPs. The strong association of the del-In9 with PSP was again verified in both the UK and US cohorts (p = 1.14×10−5, 4.021×10−8, respectively; table 3). The same trend was observed in CBD but the difference was not significant, possibly because of the small sample size. No evidence of association was found for htSNPs 8, 17, and 25 in the studies, except in the US CBD study where htSNP 17 is moderately associated (p = 0.019, allelic) (table 3). We calculated the odds ratios (OR) and their 95% confidence intervals and present values for all six htSNPs (table 3) by comparison of each minor allele verses each major allele. The H2 haplotype as defined by del-In9 is a significant protective factor. The H1 specific SNPs rs242557 and rs2471738 are highly associated with these diseases and are arguably as important for risk as the association of the extended H1 haplotype. This could particularly be the case in CBD in the light of the lack of association of del-In9 in this particular study.
Allele frequencies (F1) and p-values of single-locus association in the three studies
There is potentially a greater power to detect the contribution to association of causal variants by undertaking tests of association for the htSNP defined haplotypes rather than individual htSNPs themselves. The six htSNPs we identified capture 95% of the common haplotypic diversity of MAPT and we carried out an omnibus test of haplotype frequency differences estimated by EM between cases and controls in both the UK and US PSP groups. We found the haplotype distribution (all haplotypes >1.0%) was highly significant in the UK PSP cohort (p = 9.75×10−5, df = 19) and in the US PSP cohort (p = 7.40×10−12, df = 20) but not in CBD (p = 0.120, df = 17). In addition to the global significance of the haplotype-wide comparison, we undertook individual haplotype tests (df = 1) for significance through LRT, and derived empirical p values through Monte-Carlo methods (20 000 simulations, data not shown); we identified two common haplotypes, A and C, which were strongly associated with both UK and US PSP (table 4). Haplotype A, which derives from the del-In9 defined H2 haplotype, was the most common type in the controls and was significantly underrepresented in both PSP groups. Haplotype C, a variant of the H1 clade, was highly overrepresented in PSP. It was the commonest haplotype in PSP but not in the control groups. The most common H1 derived haplotype in the control population was not associated with either PSP or CBD. These trends were observed in CBD (table 4), though on correction for multiple comparisons no haplotype was significantly associated. In both PSP cohorts, after strict correction according to the number of tests performed, only associations of haplotypes A and C remained significant. Associated haplotypes A and C, derived from the H2 and H1 haplotypes respectively, differ by only two H1 specific htSNPs, 14 and 21, which, in addition to del-In9, also show powerful single locus effects. Haplotypes A and C do not differ by htSNPs 8 and 25, and these SNPs are not associated. The reduction in haplotype A (H2) appears almost entirely accounted for by the increase in the H1 haplotype C.
Association of common MAPT haplotypes with progressive supranuclear palsy and corticobasal degeneration
Common variation in MAPT is associated with PSP and CBD
To assess whether the significant association with PSP of any of the H1 specific htSNPs is independent of that of del-In9, we incorporated each htSNP as an additional explanatory factor to the logistic regression model of the del-In9 that serves to define the extended H1 and H2 haplotype status. We found significant association of single locus htSNPs 14, 17, and 21 (p = 9.00×10−6, 2.87×10−3 and 2.73×10−3 respectively) for the US PSP cases, htSNP 21 (p = 0.0421) for the UK PSP cases, and htSNPs 14 and 21 (p = 0.0183 and 0.0436, respectively) for the CBD cases. We probed for effects of haplotypes on subsets of htSNPs, again entering the extended haplotype (H1 and H2 status, defined by the del-In9) as an explanatory factor. We found highly significant differences in the distribution of haplotypes defined by three htSNPs 14, 17, and 21 in the UK and US PSP, and to a lesser extent in the CBD cases (p = 9.34×10−4, p = 9.31×10−5, and p = 0.0292, respectively). This was significant (p = 2.49×10−5, p = 1.44×10−8, and p = 0.006) in UK PSP, US PSP, and CBD, respectively, when the extended haplotype was excluded as an explanatory factor (table 5). The haplotypes they define are associated with PSP and CBD after consideration of the del-in9, suggesting that variability of MAPT within the extended H1 clade is a risk factor in PSP and CBD. Haplotype II (A-G-T) was greatly overrepresented in each group, and the haplotype I (G-G-C) underrepresented (table 5). The SNPs 14, 17, and 21 (rs242557, rs3785883, and rs2471738, respectively) are H1 specific SNPs in MAPT—that is, variable only on the H1 background, though the haplotype I allelic combination is fixed and representative of H2 in addition to H1 derived variants.
Association of the subset of htSNP haplotypes with progressive supranuclear palsy and corticobasal degeneration
We also attempted to reanalyse the htSNP data, after removing all individuals with an H2 chromosome, thus leaving us with a biased H1H1 homozygote population. We found significant heterogeneity (p<0.05) in both the control groups after the removal of the H2 chromosomes, namely at rs1467967 and rs7521 in the US group and at rs242557, rs2471738, and rs7521 in the UK controls. Removal of the H2 chromosomes would therefore prevent us from performing valid “H1-only” haplotype analyses in our white cohorts. For this purpose, it would be important to extend this study in an H1-only population such as the Japanese and Taiwanese.30
DISCUSSION
To date, genetic association studies have involved the study of one or a few random polymorphisms in a gene, an approach that bears the risk of missing adjacent regions of LD within the gene that harbour variants associated with phenotype. It is therefore important that the haplotype architecture of the entire gene is considered in order to determine its association with a particular complex phenotype. In our attempt to provide insight into the basis of the well established association of MAPT with PSP and CBD, we applied the haplotype tagging approach. This protocol, which uses a minimal set of tagging SNPs to study the LD and common haplotypic diversity of the entire gene or locus, is substantially more stream lined and economical.
We first assessed the underlying LD and haplotype structure of MAPT using a high density map of genotype data from the HapMap project (http://www.hapmap.org). This involved LD analysis using genotype data for 24 SNPs that had been validated in CEPH trios. In addition, we included the del-In9 status, defining the H1 and H2 haplotypes.17 This revealed multiple distinct haplotypes based upon the H1 and H2, as defined by del-In9, with no evidence of recombination between the multiple H1 haplotypes and the H2 in the CEPH trios. The presence of multiple H1 haplotypes, inferred both by EM and resolved to phase, shows a considerable diversity within this extended haplotype. This H1 haplotype specific diversity was first suggested by Golbe and colleagues, based on microsatellite variability.31 The strict H1/H2 dichotomy and H1 diversity across MAPT and beyond has also been demonstrated in other studies.25,32 In a more recent study,33 the lack of recombination between H1 and H2 has been shown to be caused by inversion of the chromosomal region on 17q21.31 corresponding to the extended MAPT H1/H2 haplotype block that we had previously described.20
We then used association based criteria to assign a set of five haplotype tagging SNPs (htSNPs) which, together with del-In9 as a sixth biallelic tagging polymorphism, capture 95% of the common haplotype diversity in MAPT. We genotyped the six htSNPs in two PSP and one CBD case–control cohorts in order to determine if any particular haplotype had greater association with disease with the extended H1. In PSP we showed clearly that there were very strong associations of two common haplotypes—first, the significant underrepresentation of the “classical” H2 (haplotype A, table 4), and second, strong overrepresentation of an H1 derived haplotype (haplotype C, table 4). The other htSNP derived common H1 haplotype (haplotype B) showed no association in any of the groups. Some weaker associations of rare haplotypes were detected but were not consistent in both the British and American cohorts in PSP, and the significance did not remain after correction for multiple comparisons. Furthermore, it is difficult to assess the association of such low frequency haplotypes in populations of our sample size. Similar trends were observed in the small number of CBD cases (n = 44), with underrepresentation of H2 (Haplotype A; table 4) and overrepresentation of the H1 derived haplotype C (table 4). However, they were not significant, possibly because of the smaller number of CBD cases. Assuming that these findings can be confirmed in a larger CBD cohort, they suggest that causative variants in PSP and CBD may affect the same region of MAPT or perhaps even be the same variant.
Pastor and colleagues defined an extended region in LD of 1.14 Mb around MAPT that is associated with PSP and CBD.34 Within this haplotype, they similarly defined a “protective” H2 haplotype that has a significant negative association with PSP and CBD, and an H1 derived haplotype that is associated with PSP and CBD.34 Our work refines the analysis of LD, haplotype structure, and associations of the MAPT gene alone and we have demonstrated that a particular H1 derived haplotype in MAPT is highly associated with PSP.
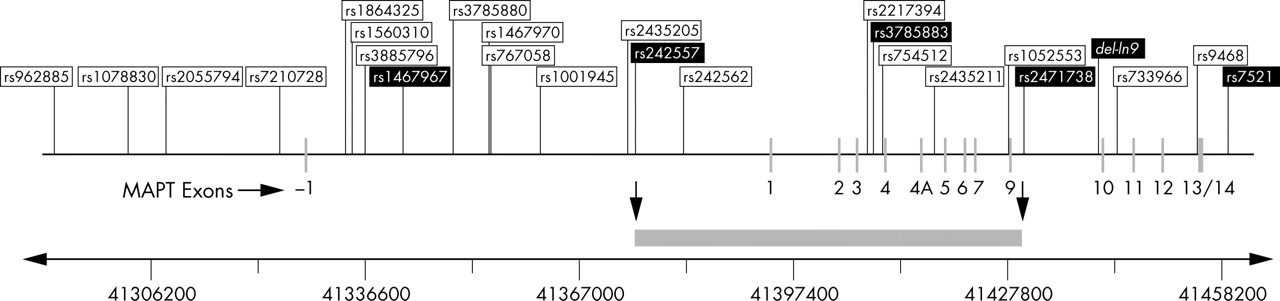
In an attempt to further minimise the candidate pathogenic domain of MAPT, we also identified particularly strong association with PSP and CBD of three-locus haplotypes based on the subset of H1 specific htSNPs, 14, 17, and 21 (table 3). These associations are independent of the extended H1 and H2 haplotypes, defined by del-In9. As indicated in fig 3, haplotypes derived from these SNPs span a minimum region from SNPs 14 (rs242557) to 21 (rs2471738) on the H1 haplotype background in MAPT. This minimum region incorporates ∼56.3 kb of sequence, from upstream of exon 1 downstream to intron 9, that could harbour potential causal variants that are in LD with these SNPs. Skipper and colleagues defined a similar associated candidate region in the 5′-half of MAPT in Norwegian Parkinson’s disease cases, thereby proposing genetic variability that could influence the alternative splicing of MAPT exons 2 and 3, or expression levels of MAPT.25 However, they carried out their analysis only on H1 homozygous individuals, having removed all H2 carriers.25 For this reason, we cannot compare findings from both studies. As explained above in Results, unbiased inclusion of the entire study cohort, irrespective of H1/H2 status, is essential in order to obtain an accurate representation of haplotype diversity in the population in question. Another study implicated an MAPT promoter haplotype in Parkinson’s disease, based not only on allelic association of the previously defined extended H1 haplotype but also on differences in transcriptional activity.35 In future studies, it would be important to compare LD and association of the MAPT locus in PSP, CBD, and Parkinson’s disease using standardised procedures, in order to determine if they share the same risk variants of the MAPT locus that contribute to disease.

{kind=link}
{kind=link}
{kind=link}
Genomic organisation of MAPT and distribution of the 25 markers used in the linkage disequilibrium and haplotype analysis in the CEPH trios. Relative positions of the promoter, coding exons, and genetic markers in the HapMap CEPH trios are to scale. Chromosome coordinates (base pairs) are according to the March 2004 build of the Human Genome Sequence (http://genome.ucsc.edu). Haplotype tagging SNPs selected for this study are indicated by the black boxes. The minimum candidate region identified in this study, in which potential causal variants may lie on the H1 background, is indicated by the grey bar.
The haplotypes we identified that confer protection, risk, or are neutral in PSP and CBD pathogenesis provide us with the basis for targeted direct sequencing strategies for MAPT. It is now clear that there are no obvious pathogenic missense or splice site mutations in MAPT in the large majority of sporadic PSP cases.17 It is more plausible that the associated SNPs in our study that confer greatest risk (SNPs 14 (rs242557) and 21(rs2471738); table 1 and fig 3) or protection (del-In9 and associated SNPs through LD; table 1 and fig 1) are in LD with variants that could cause subtle changes either in the alternative splicing or in overall expression levels. It is possible that each neuronal subgroup is dependent on a particular tau isoform profile and expression level. Aberrations in this homeostasis could affect one neuronal subgroup more than another and lead to the selective and disease specific neuronal death and tau pathology.36 Investigating correlations between candidate polymorphisms and MAPT splicing and allele specific expression—combined with the association studies described in this work—and the resulting identification of candidate variations by stringently targeted resequencing strategies in individuals carrying the haplotypes described here, could help us gain further insight into the precise nature of the role of MAPT in the molecular pathogenesis of PSP, CBD, Parkinson’s disease, and the tauopathies.
Acknowledgments
We thank the patients and their families, without whose generous support none of this research would have been possible. This work was supported by the Reta Lila Weston Trust for Medical Research, the PSP (Europe) Association (http://www.pspeur.org), the Society for PSP, USA, the Parkinson’s Disease Society, UK, the Brain Research Trust (PAS), Medical Research Council, NIH grant p50-NS40256-06 (DD, NT), The Society for PSP Brain Bank, and by the NIA/NIH Intramural Research Program. AJM is a resident research associate of the National Academy of Sciences.
Many data and biomaterials were collected from several NIA-NACC funded sites. The directors, pathologist and technicians involved include: National Institute on Aging: Marcelle Morrison-Bogorad PhD, Tony Phelps PhD, Ruth Seemann; Johns Hopkins Alzheimer’s Disease Research Center (NIA grant No AG 05146): Juan C Troncoso MD, Dr Olga Pletnikova; University of California, Los Angeles (NIA grant No P50 AG16570):Harry Vinters MD, Justine Pomakian; The Kathleen Price Bryan Brain Bank, Duke University Medical Center (NIA grant No AG05128, NINDS grant No NS39764, NIMH MH60451 also funded by Glaxo Smith Kline): Christine Hulette MD, Director; Stanford University: Dikran Horoupian MD, Ahmad Salehi MD, PhD; New York Brain Bank, Taub Institute, Columbia University (NYBB): Jean Paul Vonsattel MD; Massachusetts General Hospital: E Tessa Hedley-Whyte MD, Karlotta Fitch; University of Michigan (NIH grant P50-AG08671): Dr Roger Albin, Lisa Bain, Eszter Gombosi; University of Kentucky: William Markesbery MD, Sonya Anderson; University Southern California: Caroll A Miller MD, Jenny Tang MS, Dimitri Diaz; Washington University, St Louis Alzheimer’s Disease Research Center: Dan McKeel MD, John C Morris MD, Eugene Johnson Jr PhD, Virginia Buckles PhD, Deborah Carter; University of Washington, Seattle: Thomas Montine MD, PhD, Aimee Schantz MEd.
REFERENCES
Footnotes
-
Competing interests: none declared