Article Text

Abstract
Rett syndrome (RS) is a severe neurodevelopmental disorder that contributes significantly to severe intellectual disability in females worldwide. It is caused by mutations in MECP2 in the majority of cases, but a proportion of atypical cases may result from mutations in CDKL5, particularly the early onset seizure variant. The relationship between MECP2 and CDKL5, and whether they cause RS through the same or different mechanisms is unknown, but is worthy of investigation. Mutations in MECP2 appear to give a growth disadvantage to both neuronal and lymphoblast cells, often resulting in skewing of X inactivation that may contribute to the large degree of phenotypic variation. MeCP2 was originally thought to be a global transcriptional repressor, but recent evidence suggests that it may have a role in regulating neuronal activity dependent expression of specific genes such as Hairy2a in Xenopus and Bdnf in mouse and rat.
- BDNF, brain derived neurotrophic factor
- CDKL5, cyclin dependent kinase-like 5
- ISSX, infantile spasms syndrome, X linked
- MECP2, methyl-CpG binding protein 2
- MR, mental retardation
- ORF, open reading frame
- RS, Rett syndrome
- STK9, serine threonine kinase 9
- XLRS, X linked retinoschisis
- CDKL5
- Rett syndrome
- STK9
- X linked disorder
- intellectual disability
Statistics from Altmetric.com
- BDNF, brain derived neurotrophic factor
- CDKL5, cyclin dependent kinase-like 5
- ISSX, infantile spasms syndrome, X linked
- MECP2, methyl-CpG binding protein 2
- MR, mental retardation
- ORF, open reading frame
- RS, Rett syndrome
- STK9, serine threonine kinase 9
- XLRS, X linked retinoschisis
Rett syndrome (RS) was first described as a clinical entity in the German literature in 1966.1 Hagberg and colleagues increased awareness of the disorder in the English medical literature in 1983 with a further description of the condition in 35 girls with strikingly similar clinical features of “progressive autism, loss of purposeful hand movements, ataxia, and acquired microcephaly”.2 RS is now recognised as a panethnic disorder, and presents an ever widening clinical phenotype.3 The estimated cumulative incidence of RS in Australia is 10 per 100 000 females by the age of 12 years4 and it is considered to be the second most common cause, after Down’s syndrome, of severe mental retardation in females.5 RS is a severe neurodevelopmental disorder characterised by the progressive loss of intellectual functioning, fine and gross motor skills and communicative abilities, deceleration of head growth, and the development of stereotypic hand movements, occurring after a period of normal development. Girls with RS often develop seizures, a disturbed breathing pattern with hyperventilation and periodic apnoea, scoliosis, growth retardation, and gait apraxia.2
The association of RS with mutations in the methyl-CpG binding protein 2 gene (MECP2) was recognised in 1999.6 The MeCP2 protein is known to bind methylated CpG sequences and recruit silencing complexes that lead to compaction and silencing of surrounding chromatin. The mechanism(s) by which MeCP2 dysfunction causes RS remains somewhat of an enigma, but recent advances in our knowledge of its precise roles in neuronal and other cell types have gone a long way towards clarifying this issue. Nevertheless, diagnosis of RS remains a clinical one, as a small proportion of clinically well defined RS patients (∼5–10%) do not appear to have MECP2 mutations.
CLINICAL OVERVIEW
RS is characterised by a specific developmental profile, with the diagnosis of RS being based on a consistent constellation of clinical features and the use of established diagnostic criteria.7 More recently, refined diagnostic criteria have been proposed to clarify previous ambiguities in interpretation of clinical features8 (table 1, fig 1), and guidelines have been developed to aid researchers in being more consistent in their reporting of clinical features in RS patients.9
Revised diagnostic criteria for classical and variant RS. Derived from tables 3 and 4 in the paper by Hagberg et al.8
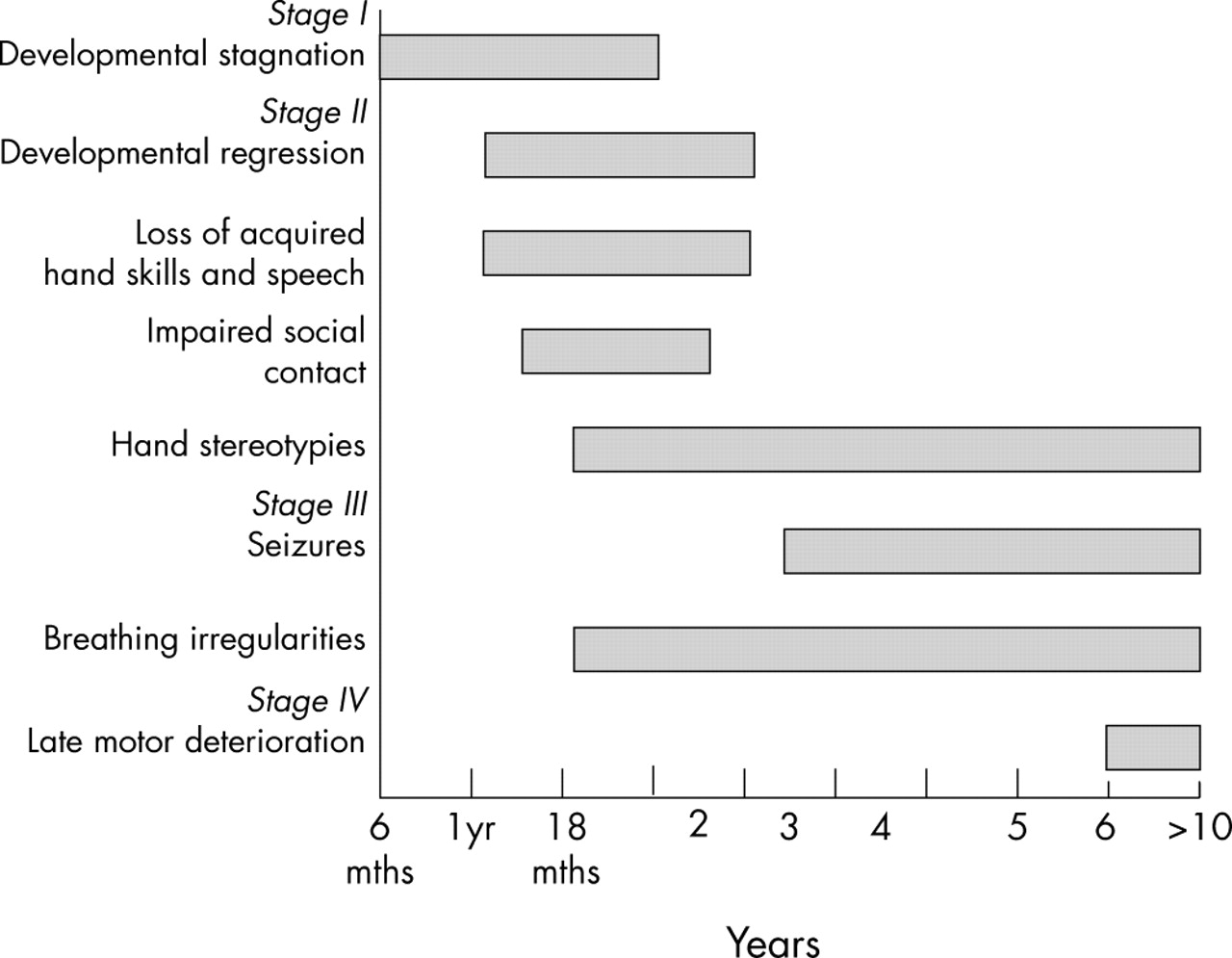
Staging system for classical Rett syndrome. Derived from Hagberg and Witt-Engerstrom.10
The diagnostic criteria for classical RS include a normal prenatal and perinatal period with normal developmental progress for the first 5–6 months of life. The birth head circumference is normal with subsequent deceleration of head growth, usually leading to microcephaly. Between 3 months and 3 years there is reduction or loss of acquired skills such as purposeful hand function, vocalisation, and communication skills. The hallmark of RS is the intense, sometimes continuous, stereotypic hand movements, which develop after the loss of purposeful hand movements. Patterns consist of tortuous hand wringing, hand washing, clapping, patting, or other more bizarre hand automatisms, during waking hours.5 A jerky truncal and or gait ataxia is another prominent feature. Supportive diagnostic criteria include breathing dysfunction, EEG abnormalities, spasticity, peripheral vasomotor disturbance, scoliosis, and growth retardation. It has been suggested that the classical RS phenotype can be described as having a four stage clinical evolution, and this has helped in discussing the natural history of the disorder with parents.10 According to the recently revised criteria,8 the clinical diagnosis of RS is excluded if there is evidence of a storage disorder, retinopathy, cataract, or optic atrophy, an identifiable metabolic or neurodegenerative disorder, an acquired neurological disorder, or evidence of perinatal or postnatal brain injury. It must not be forgotten that RS may occur coincidently with other disorders,11,12 potentially delaying the diagnosis.
With increasing experience it has become clear that females with RS may present with a much broader phenotype than originally described. A number of variants have been described, which may be more or less severe than the clinical picture seen in classical RS.13 Although initially thought to be a disorder exclusively affecting females, males with a Rett-like phenotype have been reported occasionally, including those who also have a 47XXY karyotype,14 males who are mosaic for severe mutations,15 and males who may have milder mutations.16 In addition, other non-RS phenotypes have been associated with MECP2 mutations (see below).
Management
Medical management of RS is essentially symptomatic and supportive. A multidisciplinary team approach is advocated, aimed at maximising each patient’s abilities and facilitating any skills that may be emerging. Management should include psychosocial support for the families, development of an appropriate education plan, and assessment of available community resources. Parent support groups are crucial in providing support for families. Pharmacological treatments for RS have included L-carnitine, which may lead to an improvement in patient wellbeing and quality of life,17–19 magnesium to reduce the episodes of hyperventilation,20 and melatonin to improve sleep dysfunction.21 Evaluation of the efficacy of these and other potential treatments on the horizon will require carefully constructed clinical trials, using validated instruments for measuring clinical improvements and relevant biochemical markers.
Decreasing repetitive purposeless hand movements can be achieved by the use of various arm restraints, such as soft elbow splints, and are occasionally helpful in training specific hand skills such as self feeding. These methods are also helpful in decreasing agitation and self injurious behaviour.22
Feeding problems are common in RS.23 Several factors contribute to this, including poor caloric intake secondary to swallowing difficulties and immature chewing patterns, and energy expenditure imbalances with calories used to sustain motor activities at the expense of growth. Despite a voracious appetite, some girls experience poor weight gain. This may be because the majority of girls are unable to feed themselves, and very few develop mature chewing patterns. A gastrostomy tube may be used as an alternate route to supplement nutrition. Gastro-oesophageal reflux may respond to conservative medical treatment with anti-reflux agents, thickened feeds, and positioning. Budden found that frequent small feeds during the day with added carbohydrate foods not only maintained growth and weight gain, but had a definite influence on agitation and irritability in younger girls.23
The majority of RS girls lose verbal expressive language, although some retain some speech or single word expressions. Alternative forms of communication that may be used include communication boards, technical devices, and switch activated systems. These are used for making choices and facilitate environmental access. Some girls are also able to communicate through eye pointing, gestures, body language, and hand pointing.22 These abilities need to be recognised and encouraged.
Seizure control is a common problem in the care of females with RS. A major challenge in diagnosis may be differentiating seizures from the behavioural patterns often associated with RS. Breathing irregularities such as breath holding and hyperventilation, episodes of motor activity such as twitching, jerking, or trembling, or a cardiac arrhythmia associated with a prolonged QT interval are most commonly confused with seizures. Studies using prolonged video EEG polygraphic monitoring indicate that the occurrence of seizures is overestimated. Most episodes identified by parents represent non-epileptic behavioural events. On the other hand, some actual seizures may be unrecognised by parents or occur during sleep.24 EEG telemetry and parental education may assist in identifying true seizure events.
Prolonged heart rate corrected QT values have been reported in association with RS.25,26 Prokinetic agents (such as cisapride), antipsychotics (such as thioridazine), tricyclic antidepressants (such as imipramine), anti-arrhythmics (such as quinidine, sotolol, amiodarione), anaesthetic agents (such as thiopental, succinylcholine), and antibiotics (such as erythromycin, ketoconazole) should therefore be avoided because of the possibility of precipitating electrocardiogram QT abnormalities and cardiac arrhythmias.
Scoliosis is found in approximately 65% of girls with RS. Some girls require bracing, while others require surgical intervention.22,27 Increased tone in the Achilles tendon is one of the earliest manifestations of onset of rigidity, usually followed by toe walking. It is important to maintain ambulation, and so bilateral ankle foot orthoses need to be used to prevent foot deformities, maintain foot alignment, and keep the heel cords lengthened. Physiotherapy is also required to keep the Achilles tendons stretched.22
MECP2 MUTATIONS IN RS AND OTHER CLINICAL PHENOTYPES
The MECP2 gene is located at q28 on the human X chromosome, and has been demonstrated to undergo X inactivation in mice and humans.28,29 It encodes a protein of 498 (MeCP2B or MeCP2α; encompassing exon 1 but not exon 2) or 486 amino acids (MeCP2A or MeCP2β; encompassing part of exon 2 but not exon 1), depending on the use of alternative splice variants,30,31 (figure 2A), with MeCP2B being the predominant isoform in brain. MeCP2A may predominate in other tissues, such as fibroblast and lymphoblast cells.31 There are known MeCP2 orthologues in rat, mouse, monkey, Xenopus, and zebrafish (http://mecp2.chw.edu.au/mecp2/info/MECP2_homologues.shtml), suggesting that MeCP2 has served important functions throughout vertebrate evolution, although these may be different between widely divergent species.

(A) Structure of the MECP2 gene and mRNA (adapted from Kriaucionis and Bird,30 and Mnatzakanian et al31). Alternative splicing for the A (β) isoform is shown above the gene and for the B (α) isoform below the gene. The B isoform has the highest expression in brain, and mutations specific to this isoform are sufficient to cause RS.31 (B) Structure of the B isoform MeCP2 protein sequence. The protein is 486 amino acids in size. MBD, methyl-CpG binding domain; TRD, transcription repression domain; NLS, nuclear localisation signal.
Apart from a nuclear localisation signal, MeCP2 has three functional domains (figure 2B). The methyl-CpG binding domain, at position 0–174, binds exclusively to symmetrically methylated CpGs. The transcriptional repression domain, at position 219–322, is able to recruit co-repressor complexes that mediate repression through deacetylation of core histones, with consequent compaction of DNA into heterochromatin.32,33 MeCP2 interacts with either the Sin3A/HDACI or Ski/NcoR/HDACII repression complexes to do this.34 In addition, MeCP2 also has HDAC independent silencing capabilities.35 The WW domain binding region, from residue 337 to the C terminus, specifically binds to group II WW domains of splicing factors including formin binding protein II and Huntington yeast protein C (HYPC).36 MeCP2 thus has the capability to interpret methylated CpGs differently, possibly leading to different downstream responses, depending on the context.
Following the first report of MECP2 mutations in RS patients,6 there was intense mutation screening of cohorts of patients, and attempts to draw phenotype–genotype correlations, with conflicting results.37–43 Several mutation databases have also been developed (http://homepages.ed.ac.uk/skirmis/;http://mecp2.chw.edu.au/).44 There is some agreement that missense mutations are generally milder than nonsense mutations, that mutations in the methyl binding domain are often more severe than those in the transcription repression domain, and that skewing of X inactivation can modulate the severity of the disorder. In addition, studies of a number of patients with the same mutation have made it possible to draw firm conclusions as to the severity of specific mutations.43,45,46 However, variation may still be seen between patients with the same mutation and apparently random X inactivation, suggesting that other mechanisms may also modulate the clinical severity, with differences in upstream regulators or downstream targets of MeCP2 being possible candidates.
The association of skewing of X inactivation with RS has long been a subject of debate. An increased incidence of skewing has been reported by some authors,41,47–49 but not others.50–54 Skewing has been investigated in a limited number of regions in a small number of RS brain samples,55 and these authors found random inactivation in 10/10 samples that were informative for the assay. However, it has been demonstrated that both lymphocytes56 and neuronal cells57 expressing mutant MeCP2 are at an in vitro growth disadvantage. In addition, two different MeCP2 null mouse models showed uniform distribution of MeCP2 in the brains of heterozygous females, but skewed X inactivation favouring the wild type allele.58–60 Young and Zoghbi57 also identified regional skewing in the brains of mice carrying the common 308X mutation, and we have demonstrated that there may be regional variation in human RS brains.61 Furthermore, it has been demonstrated that skewing in the brains of mutant mice correlates with the phenotypic outcome, with the chance of manifestation of certain symptoms decreasing with increasing expression of the wild type allele.57 These studies, combined with previous studies of X inactivation in RS patients, strongly suggest that skewing plays a significant role in modulating phenotypic severity.
Prior to the identification of the MeCP2B (MeCP2α) isoform, mutation detection efforts had focused on PCR based approaches to screening exons 2, 3, and 4. More recently, it has been recognised that up to 15% of apparently MECP2 mutation negative individuals have large deletions (spanning kilobases) that would be missed by this approach.62–65 In addition, the identification of the new MeCP2 isoform has raised the speculation that some individuals may have mutations in exon 1,31 although in our experience this appears to be an uncommon event (unpublished observations).66 Using a combination of PCR based screening approaches for the MECP2 coding regions and one or other method to screen for large deletions, it has been suggested that pathogenic MECP2 defects will be identified in up to 90–95% of RS cases.63 A remaining challenge is to identify the genetic defect in the remaining 5–10% of cases, with possible regions of interest including the MECP2 promoter region, the blocks of highly conserved sequence within the very large 3′ untranslated region,67 or novel genes (see below).68,69
Interestingly, the vast majority of single nucleotide changes in the MECP2 gene are the result of C→T transitions at CpG hotspots.41,70,71 The general restriction of RS to females and the very low recurrence rate72 appear to be the result of a large proportion of these mutations arising via spontaneous deamination of 5-methylcytosine to thymine in the heavily methylated male germ cells.73–75
A large degree of phenotypic variability is seen in both RS patients and people with MECP2 mutations, ranging from classical RS to normal individuals with protective skewing of X inactivation. The phenotype associated with MECP2 mutations has broadened even further to include males with severe neonatal encephalopathy,70,76, males from X linked mental retardation pedigrees,77–81 PPM-X syndrome,82 Angelman syndrome,83,84 and infantile autism.85
FUNCTIONAL CONSEQUENCES OF MECP2 MUTATIONS
MeCP2 was initially thought to function as a global transcriptional repressor, but the specifically neuronal phenotype associated with mutations, and the apparent lack of global gene dysregulation in cell lines from RS patients,86,87 and in brains from human RS patients88 and RS mouse models,87 do not support this suggestion.
Recent studies have begun to identify specific MeCP2 targets. In Xenopus, MeCP2 inhibits the expression of xHairy2a, which itself is a target of the Notch/Delta signalling pathway.89 With reduction of MeCP2 activity, xHairy2a expression is increased, and this leads to inhibition of primary neurogenesis.89 It will be interesting to study whether MeCP2 plays a role in other Notch regulated aspects of neuronal maturation, including dendritic branching, which is known to be abnormal in the RS brain.
Studies of MeCP2 expression in primate prefrontal cortex demonstrate increases during development, with expression expanding from the deeper cortical layers and subplate in 110 day embryos, to robust expression throughout the prefrontal cortex in adult monkeys.90 Moreover, increased MeCP2 expression appears to be associated with neuronal maturation, particularly in the hippocampus, cortex, and cerebellum, the brain regions primarily affected in RS.91,92 These combined expression data suggest that MeCP2 has specific functions in neuronal cells of the central nervous system. Indeed, Aber et al93 demonstrated that MeCP2 localises both to the postsynaptic compartments of neuronal cells and to the nucleus, suggesting that MeCP2 links the regulation of transcription to synaptic activity.
Two recent papers have identified one possible MeCP2 target in this pathway in mammals.94,95 These authors found that MeCP2 binds specifically to BDNF (brain derived neurotrophic factor) promoter III in rat and promoter IV in mouse, respectively, thereby repressing BDNF transcription in resting neuronal cells. Membrane depolarisation of neuronal cells led to calcium dependent phosphorylation and release of MeCP2 (fig 3), suggesting that MeCP2 may regulate the transcription of activity dependent genes in neuronal cells,94 which is important in synapse development and neuronal plasticity.
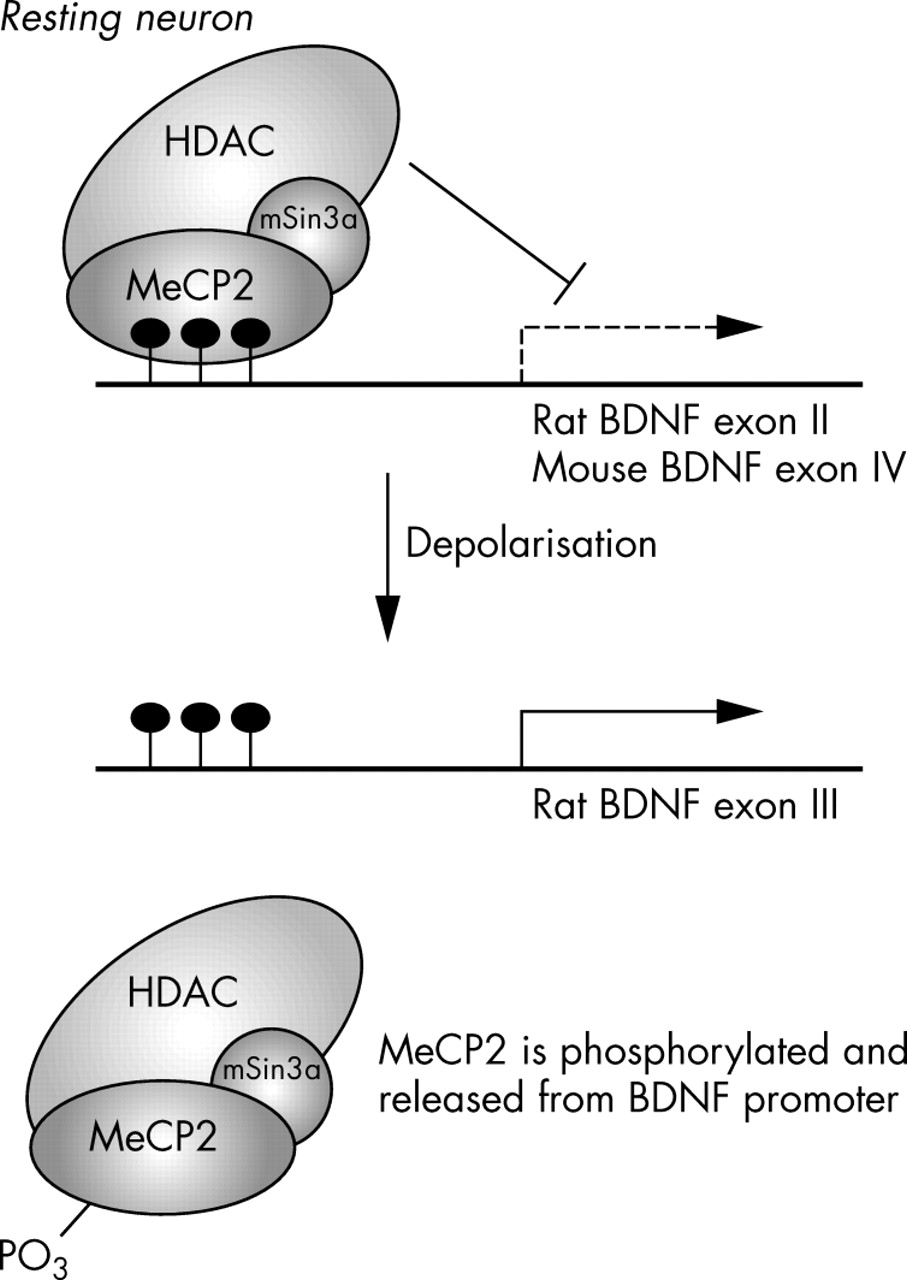
Proposed model for MeCP2 regulated expression of BDNF. In the resting neurone, MeCP2 binds the BDNF promoter, repressing its activity. Following neuronal depolarisation, MeCP2 is phosphorylated, perhaps by CDKL5, leading to its displacement from the BDNF promoter, and thus allowing BDNF activation.
Deletion of the C terminus of MECP2 encompassing the WW domain binding region occurs commonly in RS patients, and is associated with loss of binding to the splicing factors FBP11 and HYPC.36 Preliminary evidence from our laboratory suggests that cell lines carrying C terminal deletions may shift towards recruiting smaller protein complexes on binding unmethylated or partially methylated CpG rich regions (data not shown). As these C terminal deletions do not disrupt the MBD or the TRD, the pathogenesis in these cases may be due to loss of the WW domain binding region, and hence, loss of the proteins that normally interact with this domain. These findings are yet to be confirmed in brain and neuronal cells,36 but if they are, it raises the interesting possibility that MeCP2 is involved in splicing, or that WW binding domains may be found in specific neuronal targets of MeCP2.
CDKL5: A SECOND RS GENE?
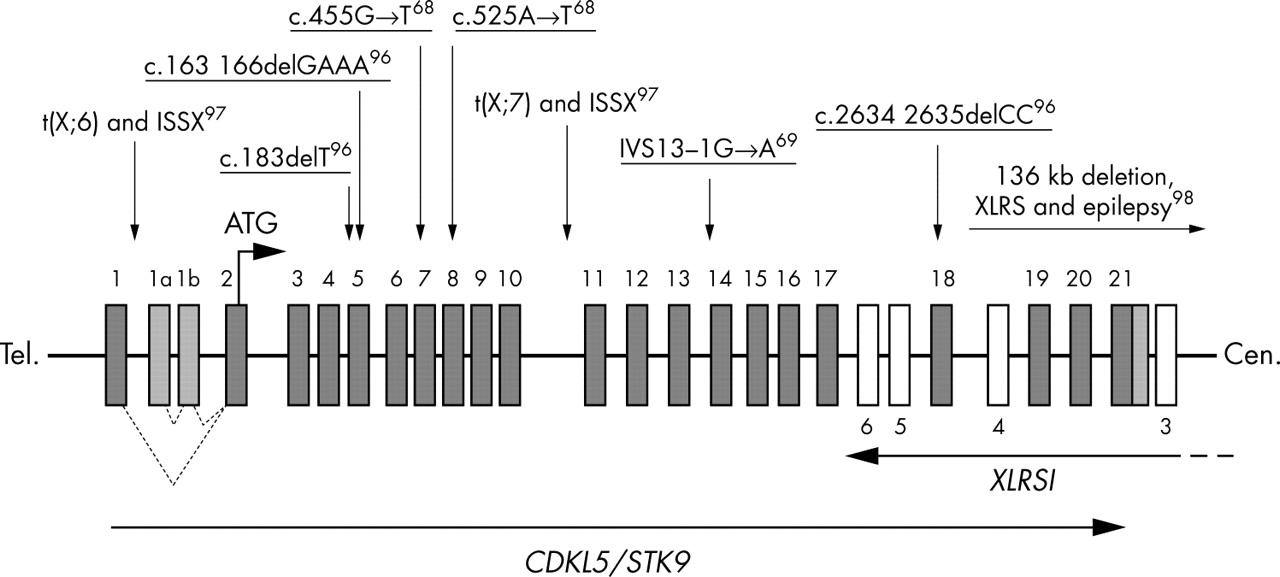
The apparent lack of MECP2 mutations in a small proportion of clinically well defined RS cases suggests the existence of at least one other RS locus.41,71 Three recent reports,68,69,96 identified mutations in the gene for cyclin dependent kinase-like 5 (CDKL5; OMIM #300203; also known as serine threonine kinase 9 (STK9)) in patients who had been diagnosed with atypical RS, supporting the existence of genetic heterogeneity in RS. Somewhat similar to the MECP2 involvement in RS, mutations affecting CDKL5 appear to yield clinical outcomes of varying severity. In addition to two female patients with infantile spasms syndrome, X linked (ISSX) and X;autosome translocations interrupting the CDKL5 gene who were described by Kalscheuer et al,97 RS patients with early onset seizures (seven cases), one girl with an autistic disorder and intellectual disability, and a boy with severe, early infantile onset neurological disorder have CDKL5 mutations.68,69,96 Additionally, one male patient with X linked retinoschisis (XLRS) and a large deletion involving at least the last coding exon of the CDKL5 gene had seizures, which are otherwise not associated with XLRS.98 In this case it is likely that the seizure phenotype was due to deletion of the CDKL5 gene, but other gene contributions cannot be excluded. In summary, there are currently nine published (fig 4) and at least two unpublished RS cases (personal communication: Dr H Archer, Dr J Evans, Professor A Clarke, Department of Medical Genetics, University of Wales College of Medicine, Cardiff, Wales) with mutations in, or involving the CDKL5 gene. From among these, eight cases have been diagnosed with the atypical, early seizure variant of RS, suggesting that this may be the predominant phenotype caused by CDKL5 mutations. Although still very tentative, some speculations about the CDKL5 mutation genotype/phenotype correlation can be drawn. While protein truncating mutations at the N terminal end97 cause a severe, early onset phenotype of infantile spasms (phenotype overlapping with that of ISSX as caused by the Aristaless (ARX) gene mutations99), truncations more towards the C terminal end cause atypical RS in girls, a severe neurological disorder in boys, or autistic disorder with intellectual disability. Tissue specific skewing of X inactivation, genetic background, and as yet unknown environmental factors may contribute to the phenotypic variability, as revealed in at least one case of discordant monozygous twins.69

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Exon structure of the CDKL5 (STK9) gene and its overlap with the XLRS1 gene. Published mutations involving the CDKL5 gene are indicated with arrows. Mutations found in patients with early seizure variant of RS are underlined. The position of two translocation breakpoints (associated with ISSX) and a deletion involving both XLRS1 and CDKL5 genes is also indicated. All known exons of the CDKL5 gene (solid boxes) and some of the XLRS1 gene (empty boxes) are indicated and numbered.
CDKL5 is a putative serine/threonine kinase of unknown function.100,101 The connection between CDKL5 and MeCP2, if there is one, and the mechanism by which they produce overlapping phenotypes, will be important questions for future studies to address. CDKL5 mutations appear to be associated with severe, early onset seizures in particular, and thus CDKL5 gene screening should be considered in atypical, MECP2 negative RS cases. Whether ARX tested negative cases with infantile spasms, or autism spectrum disorder cases with intellectual disability and early onset seizures, should also be considered remains to be investigated.
CONCLUSIONS
Recent advances in our knowledge of MECP2 splicing, its specific targets, and the phenotypes and expression in mutant mice have greatly contributed to our understanding of the aetiology of RS. Studies in cell lines and mouse models have confirmed the contribution that skewing of X inactivation can make to phenotypic variability.56–58 Identification of Hairy2a89 and BDNF94,95 as bona fide targets of MeCP2 has given glimpses of the mechanisms by which MECP2 mutations lead to RS, and have enhanced the understanding of the functions of MeCP2, which no longer appears to be the global transcriptional repressor it was once assumed to be.102 Rather, it may function to regulate the dynamic expression of neuronal genes and the formation of new synaptic connections in response to electrical signalling, thereby explaining the specifically neuronal phenotype associated with MECP2 mutations. Moreover, some of the phenotypic variability observed in RS has been confirmed to result from genetic heterogeneity. Several reports have identified mutations in CDKL5 in specific atypical RS variants associated with early onset seizures. It remains to be determined whether this or other genes are responsible for a significant proportion of atypical RS cases or other neurological phenotypes.
While much has been learnt about RS since its first description over 30 years ago, the pathogenesis of this important and intriguing disorder is still not well understood. To fully explain the RS disease phenotype will require the identification of further gene targets for MeCP2 and CDKL5, particularly those predominantly active in the central nervous system.
Such studies would greatly enhance the understanding of normal brain development and function, and provide opportunities for the development of targeted therapeutic interventions for RS, possibly in the presymptomatic stage or early in the evolution of the disorder.
REFERENCES
Footnotes
-
Competing interests: none declared