Article Text

Statistics from Altmetric.com
Neurofibromatosis type 1 (NF1) is a common autosomal dominant genetic disease. In recent studies on the neurofibromatosis type 1 (NF1) gene neurofibromin, splicing abnormalities were seen in 30-50% of cases when RNA taken from cell lines was analysed.1,2 Unlike mutations that alter critical amino acids or generate premature stop codons, splicing abnormalities can be very hard to predict from sequence analysis alone. Apart from the two base pairs 5′ and 3′ of each exon, few of the nucleotides in regions critical for splicing are absolutely conserved. As a consequence, it can be very difficult to conclude that a sequence variation found in a patient will alter splicing and so represents a pathogenic mutation.
Key points
-
Abnormalities of pre-mRNA splicing represent an important mechanism by which gene mutations cause disease.
-
Effects on splicing can be predicted from genomic DNA sequence analysis if mutations alter highly conserved canonical splicing signals. However, it is extremely difficult to predict the effects of changes in intronic and exonic sequences not obviously involved in the splicing process.
-
We present here an efficient and simple test using genomic DNA to construct a minigene and analyse the effect on splicing of sequence variations.
-
Using this system, we describe the identification of one such sequence variation in the NF1 gene as the disease causing mutation. The aberrant splicing can then be rescued by coexpression of an altered U1 snRNA that restores normal base pairing.
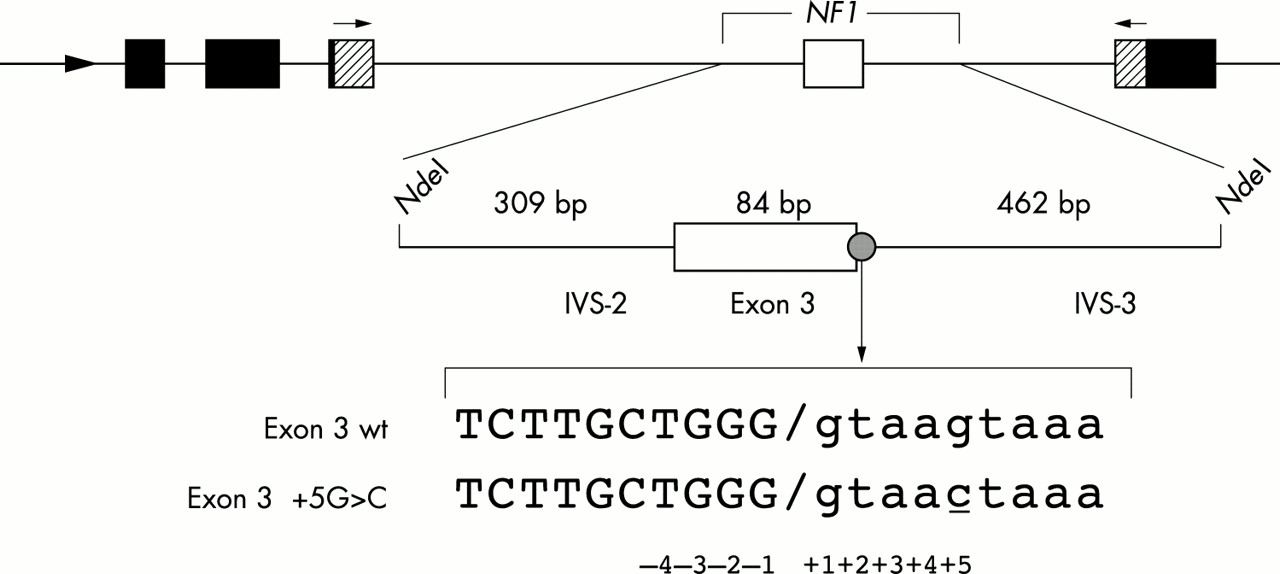
This difficulty is well illustrated by a family with NF1 in which we recently identified a sequence variation. The three generation family is from the UK and meets NIH diagnostic criteria. The index case, at the age of 82, has classical features of NF1 including multiple café au lait macules, neurofibromas, and axillary and inguinal freckling. Her son was similarly affected and died in a road traffic accident. Her granddaughter additionally had macrocephaly and died aged 31 from a malignant nerve sheath tumour affecting the coeliac axis. Through sequencing all exons in the neurofibromin gene, as well as 50 base pairs of 5′ and 3′ intronic flanking sequence, only a single nucleotide change was found, consisting of a substitution of a cytidine for a guanosine 5 bp downstream of exon 3 (exon 3+5 G>C, fig 1). This variation was seen in the proband and the granddaughter and has not been found in 100 normal chromosomes (Mattocks et al, unpublished observations), consistent with it representing the disease causing mutation. The vicinity of the nucleotide change to the 5′ splice site of intervening sequence (IVS) 3 (figs 1 and 2A) suggests that it may interfere with the splicing of exon 3. However, there are examples of wild type sequences similar to the exon 3+5 G>C mutation in which the corresponding exon is spliced efficiently; for example within the normal NF1 gene IVS-1 and -7 share identical –1 to +5 sequence with the mutated IVS-3. We were therefore unable to predict on the basis of sequence analysis alone that this sequence change will interfere with splicing and cause NF1, and a direct analysis of exon 3 splicing assay was therefore required in this family.

Schematic representation of the α-globin fibronectin NF1 exon 3 minigene (black, shaded, and white boxes respectively, with intervening sequences (introns) shown as lines). The mutation is shown with exonic sequence in upper case and intronic sequence in lower case.

{kind=link}
{kind=link}
(A) RNA products generated by the splicing assay. Two products are seen on agarose gel electrophoresis, with the 239 bp band representing an RNA product lacking exon 3 and the 323 bp band a product including exon 3, as indicated schematically on the right of the gel. Note that the C>G minigene excludes exon 3, while the WT gene includes this exon. Splicing is rescued (that is, exon 3 is included) by the coexpression within the minigene expressing cells of a U1 snRNA altered to be complementary to the mutation containing sequence (exon 3 G>C/C>G-U1) but not by expression of the WT-U1 snRNA (exon 3 G>C/WT-U1). (B) Normal base pairing between the 3′ end of exon 3 and WT-U1 snRNA. :Indicates wobble association. The nucleotide change observed in the patient (G>C at the +5 position) reduces base pairing between the RNA and the U1 snRNA as shown. (C) The variant snRNA (C>G-U1) changed to complement the nucleotide change seen in the family; note the restoration of appropriate base pairing.
METHODS
Hybrid minigene constructs
Human genomic DNA was amplified from normal and mutated (IVS 3 +5 G>C) NF1 exon 3 to generate a fragment that contained the exon 3 along with 309 bp of 5′ and 462 bp of 3′ intronic flanking sequence using the following oligonucleotides: 5′ GGAATTCCATATGTCAAGATTCTGGTACAGGTC 3′ sense; 5′ GGAATTCATATGTCTCAAGGTAACATCTATCC 3′ antisense.
Both oligonucleotides carry an NdeI restriction enzyme site in their 5′ ends, used to clone the product into a modified version of the α-globin-fibronectin-EDB minigene in which the alternatively spliced EDB exon has been removed to generate a site for the insertion of exons under study.3
Analysis of the hybrid minigene expression
The splicing assay was performed by transfecting 0.5 μg of each minigene plasmid into 3 × 105 Hep3B cells with Qiagen Effectene transfection reagents. RNA extraction and RT-PCR analysis was performed as described previously,5 using primers complementary to sequences in the flanking fibronectin exonic sequence.
In the experiment designed to rescue splicing defects, we also expressed a variant snRNA C>G-U1, complementary to the nucleotide change observed in the patient. This was generated by replacing the sequence between the sites BclI and BglII in the pGU1 plasmid4 (which contains a WT snRNA-U1 clone) with the oligonucleotides: 5′ GATCTCATAGTTACCTGGCAGGGGAGATACCAT 3′; 5′ GATCATGGTATCTCCCCTGCCAGGTAACTATGA 3′.
RESULTS AND DISCUSSION
Previous studies examining the effects on splicing of NF1 mutations have analysed RNA extracted from individual patients to confirm possible splicing defects. This is, however, a laborious technique requiring a potentially difficult RNA extraction from a cell line or tissue, and complicated by sources of variability, such as splice site leakiness in some tissues and lower levels of expression of mRNA from the mutant alleles. However, in many cases the necessary further samples may be hard to obtain or unavailable for the laboratory performing the sequence analysis. As an alternative technique, using genomic DNA and circumventing these problems of variability or the need for further sampling, we took advantage of a simple minigene splicing assay. In this assay individual exons are inserted, along with flanking intronic sequences, into a fibronectin (Fn) minigene containing upstream and downstream intronic and exonic sequences sufficient to allow splicing (fig 1). We have shown previously how this minigene system can be used to identify sequences required for the correct regulation of alternative splicing in the cystic fibrosis (CFTR) gene,3 and also to identify abnormalities of splicing of the ATM gene as a disease mechanism in ataxia-telangiectasia.4 Here we amplified genomic DNA comprising exon 3 and flanking intronic sequences (as shown in fig 1) from the potentially abnormal NF1 gene in this family and inserted this into the minigene. Following transfection and expression of the construct in Hep3B cells, the mRNA produced by the cells was analysed for splicing pattern by RT-PCR. As shown in fig 2A, the exon 3 +5 G>C mutation dramatically affected pre-mRNA processing, causing exon 3 to be completely skipped.
To confirm the role of this mutation further, we constructed and expressed in the cells a U1 snRNA complementary to the mutation observed in the patient. One of the early events in the process of intron removal from mRNA precursors involves recognition of the 5′ splice site by U1 small nuclear ribonucleoprotein (snRNP). This recognition involves complementary base pairing, and the substitution of the guanosine by a cytidine in position +5 of IVS-3 lessens the degree of U1 snRNA base pairing with the 5′ splice site (fig 2B). Altering the complementary cytidine to guanosine in the U1 snRNA will restore normal base pairing (fig 2C) and, as predicted, coexpression of this altered U1 snRNA with the minigene carrying the mutation resulted in rescue of exon 3 splicing (fig 2A). In contrast, expression of WT U1 snRNA does not rescue splicing (fig 2A), showing that this does not simply result from increased levels of U1 snRNA. This experiment proves that the exon 3+5 G>C variation is a disease causing mutation that induces aberrant skipping of exon 3.
As techniques for the identification of sequence variation become faster and cheaper, the distinction between polymorphisms and pathogenic mutations will be an increasing challenge. The assay we have used here in the analysis of one NF1 family is a potentially valuable tool for the identification of those mutations that cause splicing defects. No RNA or further samples are required from the patient, making it feasible to carry out this further step in the molecular genetic diagnostic laboratory as part of the analysis of the DNA sample provided by the referring clinicians. In addition, the minigene system effectively recreates within the cell line the splicing defect of each patient, so facilitating further studies on the relationship between genotype and phenotype in this disease.
Acknowledgments
The first two authors contributed equally to this work. This work was supported by Telethon Onlus Foundation (Grant E1038 awarded to FEB), a Research Training Fellowship from Action Research (Grant SF1001 awarded to DB), and a Wellcome Trust Research Leave fellowship (awarded to CffC).