Article Text
Research Article
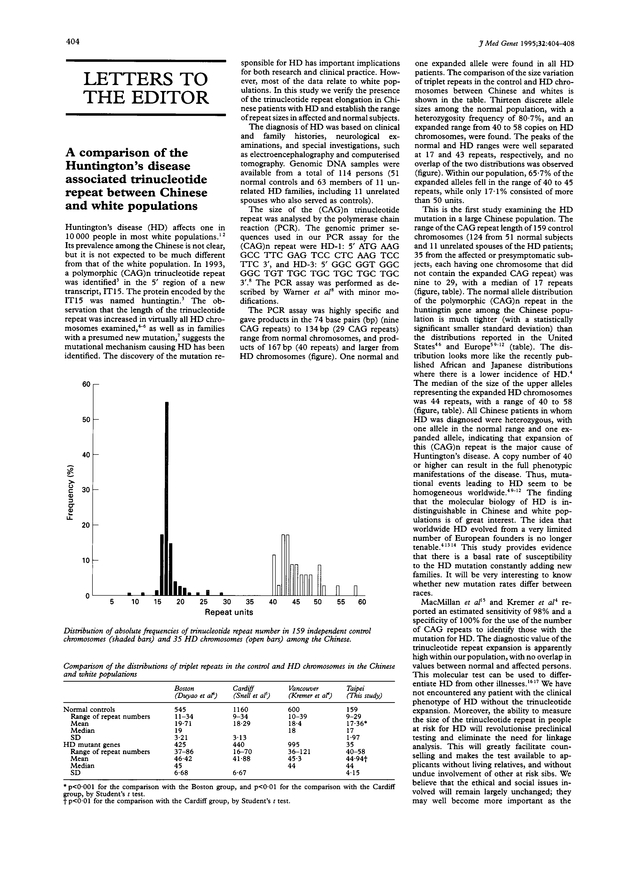
A comparison of the Huntington's disease associated trinucleotide repeat between Chinese and white populations.

Statistics from Altmetric.com
This is a PDF-only article. The first page of the PDF of this article appears above.