Article Text

Abstract
Using array comparative genome hybridisation (CGH) 41 de novo reciprocal translocations and 18 de novo complex chromosome rearrangements (CCRs) were screened. All cases had been interpreted as “balanced” by conventional cytogenetics. In all, 27 cases of reciprocal translocations were detected in patients with an abnormal phenotype, and after array CGH analysis, 11 were found to be unbalanced. Thus 40% (11 of 27) of patients with a “chromosomal phenotype” and an apparently balanced translocation were in fact unbalanced, and 18% (5 of 27) of the reciprocal translocations were instead complex rearrangements with >3 breakpoints. Fourteen fetuses with de novo, apparently balanced translocations, all but two with normal ultrasound findings, were also analysed and all were found to be normal using array CGH. Thirteen CCRs were detected in patients with abnormal phenotypes, two in women who had experienced repeated spontaneous abortions and three in fetuses. Sixteen patients were found to have unbalanced mutations, with up to 4 deletions. These results suggest that genome-wide array CGH may be advisable in all carriers of “balanced” CCRs. The parental origin of the deletions was investigated in 5 reciprocal translocations and 11 CCRs; all were found to be paternal. Using customised platforms in seven cases of CCRs, the deletion breakpoints were narrowed down to regions of a few hundred base pairs in length. No susceptibility motifs were associated with the imbalances. These results show that the phenotypic abnormalities of apparently balanced de novo CCRs are mainly due to cryptic deletions and that spermatogenesis is more prone to generate multiple chaotic chromosome imbalances and reciprocal translocations than oogenesis.
Statistics from Altmetric.com
Once it was found that the dystrophin (DMD) gene is interrupted in female patients with Duchenne muscular dystrophy (DMD) carrying Xp21/autosome balanced translocations,1 it became obvious that abnormal phenotypes, present in 6.1% of translocation carriers,2 might be caused by the breakage of dosage-sensitive genes or by the separation of the gene from its cis regulatory elements, leading to its aberrant expression. Although in translocations associated with contiguous gene syndromes, deletions at the breakpoint had never been excluded,3 the idea that the abnormal phenotype in reciprocal balanced translocations was caused by the direct breakage of a dosage-sensitive gene has become predominant. In addition to the breakage of genes responsible for autosomal dominant phenotypes, balanced translocations could unmask a mutation in the second allele causing an autosomal recessive phenotype or could cause the disruption of an imprinted gene causing an apparent imprinting disorder.4 5
Complex chromosome rearrangements (CCRs) are structural chromosome anomalies involving >2 chromosomes or >2 breakpoints. In the literature, CCRs have been reported with varying numbers of breakpoints, from the simplest, with 3 breakpoints, to the most complex, with ⩾8 breakpoints.6–8 Conventional cytogenetics is of limited use in determining whether a CCR is balanced or unbalanced. The finding that 23% of the CCRs, although apparently balanced, have been ascertained among individuals with multiple congenital anomalies and/or mental retardation, and that among those with de novo occurrence, more than half have been found in individuals with phenotypic abnormalities,8 suggests that imbalances may be a common finding. Detailed flurorescence in situ hybridisation (FISH) studies of translocation breakpoints have reported the existence of cryptic deletions near translocation breakpoints.9 The application of high resolution platforms to detect copy number changes in carriers of apparently balanced translocations is now revealing that conventional cytogenetics may have resulted in errors, by overlooking more complex situations. Several reports describing either single cases or small series of apparently balanced reciprocal translocations and complex rearrangements have revealed unexpected complexity and instability of the human genome.10 11 In particular, the complex nature of constitutional de novo apparently balanced translocations in patients with abnormal phenotypes is surprising.10 12
In this paper we systematically analysed 59 rearrangements involving both reciprocal translocations (27 found in patients with abnormal phenotype, and 14 in fetuses, all but two with normal echographic findings) and complex rearrangements (13 in patients with abnormal phenotype; 3 in fetuses with normal echographic findings; 2 in women who had experienced repeated spontaneous abortions) for the presence of cryptic chromosome imbalances.
METHODS
Patients
A brief description of the phenotypes of the patients with reciprocal translocations and the cytogenetic interpretation made by conventional investigation are reported in table 1, and table 2 shows the conventional karyotype in 14 prenatal cases. Patients 17 and 22 had been previously published.13 14
Table 3 gives a brief description of the phenotypic abnormalities of the patients with CCRs, together with their karyotype definition as stated by conventional cytogenetics. Table 4 summarises the conventional karyotype found in the two women with normal phenotypes (patients 55 and 56) who had repeated spontaneious abortions, and table 5 shows a conventional karyotype in the three prenatal cases (patients 57, 58 and 59) investigated for advanced maternal age. Patients 47, 54, and 49 have been previously published.6 15 16
FISH analysis and genotyping
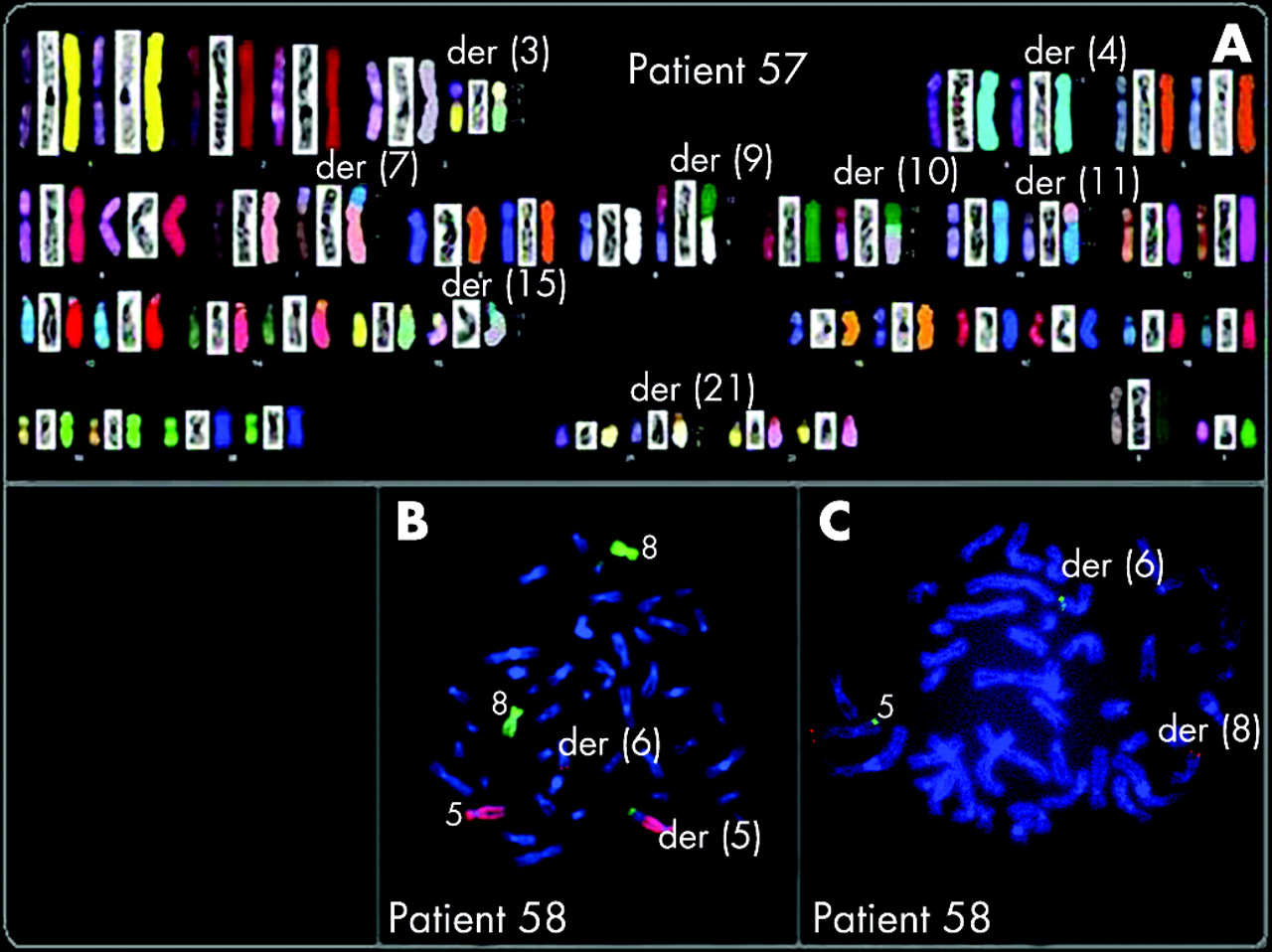
In all CCR patients except four (two already published (patients 49 and 54), patient 56 and patient 59 (prenatal patient)), FISH with whole chromosome painting probes (Chromoprobe Multiprobe System; Cytocell Technologies, Cambridge UK or Qbiogene, Irvine, California, USA) and in some patients probes for the subtelomeric regions (To Tel Vysionand Multi-color DNA Probe Mixtures; Vysis, Chicago, Illinois, USA) were applied so to better define each rearrangement. In patient 44, multicolour FISH was performed using the 24 Xcyte mFISH DNA Probe Kit, following the manufacturer’s instructions (Metasystem, Hamburg, Germany). In patient 57, a spectral karyotyping (SKY) was performed using the protocol previously reported.17 Genotyping of polymorphic loci on DNA from the the probands and parents was performed by amplification with primers labelled with fluorescent probes (ABI 5-Fam and Hex) followed by analysis on an ABI 310 Genetic Analyzer (Applied Biosystems, Foster City, California, USA).
FISH analysis of patients 43 and 50 was performed following conventional protocols as previously described.18 Bacterial artificial chromosome (BAC) clones were selected from the human library RPCI-11 according to the UCSC Human Genome Assembly (freeze May 2004) and provided by the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk/). BAC DNA was labelled with biotin and digoxigenin using nick translation. The probes were visualised using fluorescein isothiocyanate–avidin and the chromosomes were made fluorescent by 4',6-diamidino-2-phenylindole. Hybridisations were analysed with an epifluorence microscope, and images captured with a PowerGene FISH System (PSI).
DNA paternity testing
Paternity was tested in patients 43, 45–47, 49–50, 52 and 57 after informed consent. From each sample of the trios (alleged father, mother and child), 10 ng DNA was amplified by PCR, using the commercial AmpFLSTR Identifiler PCR Amplification Kit (Applied Biosystems, Alabama, USA). This product amplifies in a multiplex PCR reaction 15 short tandem repeat (STR) loci (D8S1179, D21S11, D7S820, CSF1PO, D3S1358, TH01, D13S317, D16S539, D2S1338, D19S433, vWA, TPOX, D18S51, D5S818, FGA) and the sex-typing marker amelogenin. The samples were amplified through 28 PCR cycles according to the manufacturer’s specifications and 0.2 ml of each amplified sample was analysed by capillary electrophoresis (ABI Prism 310 Genetic Analyser; Applied Biosystems). Alleles were scored using the Genotyper Software version 2.5.2 (Applied Biosystems). The statistical calculation used to evaluate the weight of the evidence for a non-excluded man was the probability of paternity (W).
Molecular karyotyping
Molecular karyotyping was performed in all patients using Agilent Technologies Array CGH Kits (Santa Clara, CA). Parents of probands showing cryptic deletions were also analysed to exclude that the imbalance was inherited. These platforms are 60-mer oligonucleotide-based microarrays that allow genome-wide survey and molecular profiling of genomic aberrations with a resolution of ∼100 kb (kit 44B) and ∼20 kb (kit 244A). Aliquots of 500 ng of DNA from patients, parents and same-sex reference samples (Promega, Madison, Wisconsin, USA) were double-digested with RsaI and AluI for 2 hours at 37°C. After heat inactivation of the enzymes at 65°C for 20 minutes, each digested sample was labelled by random priming (Agilent Technologies) for 2 hours using Cy5-dUTP for patient/parent DNAs and Cy3-dUTP for reference DNAs. Labelled products were column-purified. After probe denaturation and pre-annealing with 50 μg of Cot-1 DNA, hybridisation was performed at 65°C with rotation for 24 hours (44B) or 40 hours (244A). After two washing steps, the array was analysed using an Agilent scanner and Feature Extraction V.9.1 software. A graphical overview of the results was obtained using CGH Analytics V.3.4.27 software.
We also used Agilent customised arrays with a very high resolution (<1 kb in most patients) to narrow down the breakpoints in seven patients (42, 43, 45, 47, 49, 50 and 51) with one or two deletions. Customised arrays were designed using eArray software (http://earray.chem.agilent.com/) and probes were selected from those available in the Agilent database. We then analysed all the breakpoint sequences with self chain and segmental duplication repeats tools from UCSC (http://genome.ucsc.edu/). The tandem repeats finder tool allowed us to search for the presence of AT-rich repeats and STR sequence accumulation.
RESULTS
Reciprocal translocations: postnatal and prenatal cases
Table 6 summarises the results of array CGH showing that 11 out of 27 translocations found in patients with an abnormal phenotype were in fact unbalanced.
Figure 1 shows array CGH profiles in patients 1–11; in each figure, the deleted chromosome is detailed at the top of each profile, and the patient number below. Nine patients showed a single deletion, which occurred at one of the breakpoints in six patients (patients 1–4, 10 and 11), on chromosomes unrelated to the translocation in two (patients 5 and 9), and on a derivative chromosome but in a region different from the breakpoint in one (patient 6). Patient 8 had two deletions, one at each of the breakpoints, and patient 7 had three deletions, all on chromosomes unrelated to the translocation. The parental origin of the deletions was investigated in five patients (patients 2–7) and was found to be paternal in all of them (supplementary table 1; available at http://jmg.bmj.com/supplemental). The remaining 14 patients (28–41), all ascertained by prenatal diagnosis, were found by array CGH screening to be balanced.

Complex chromosome rearrangements
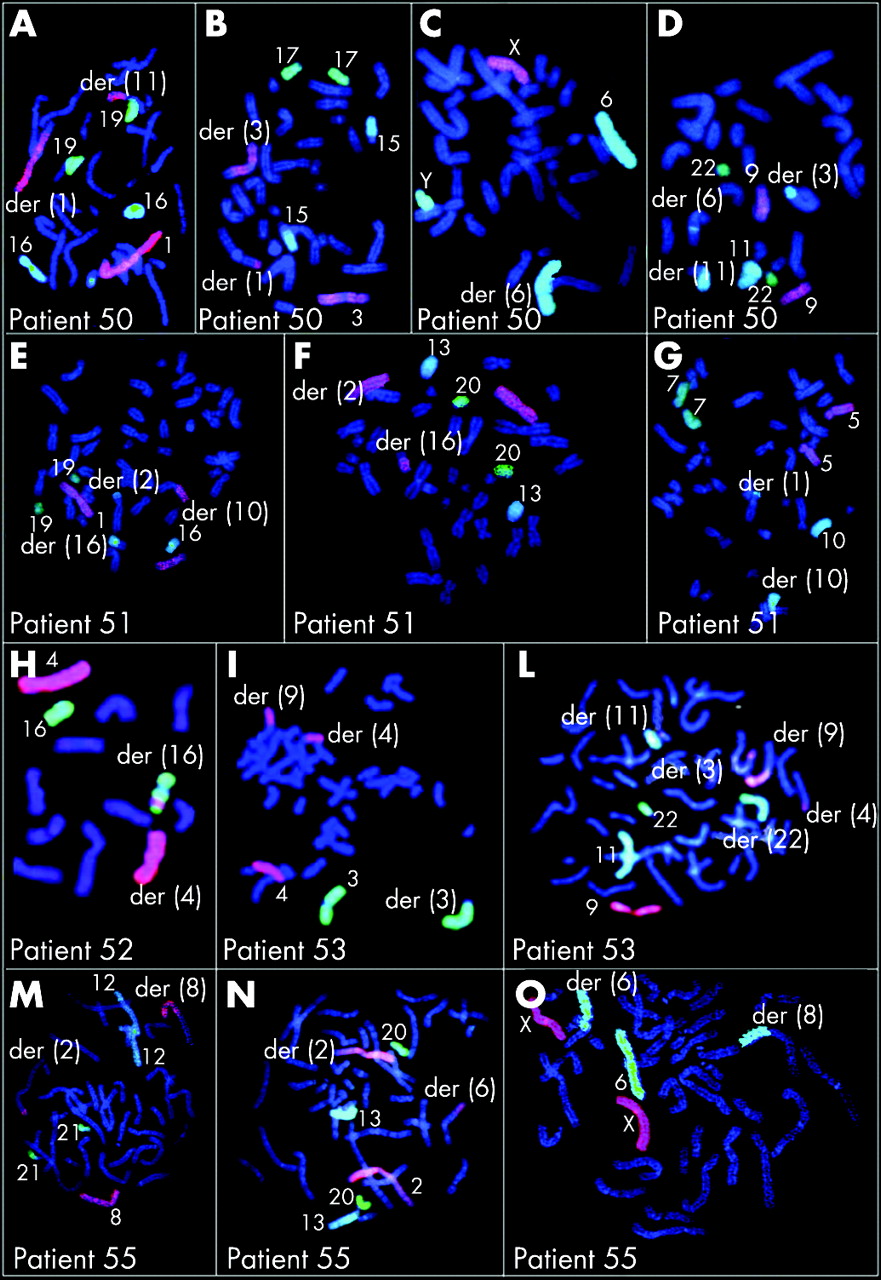
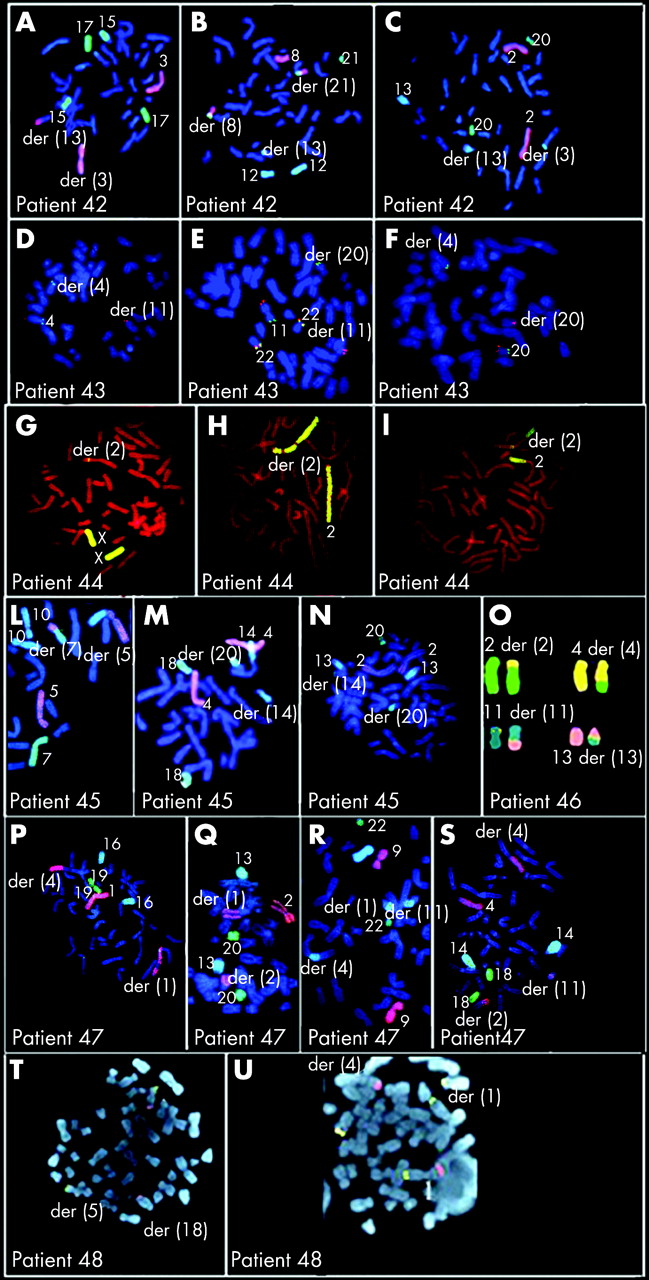
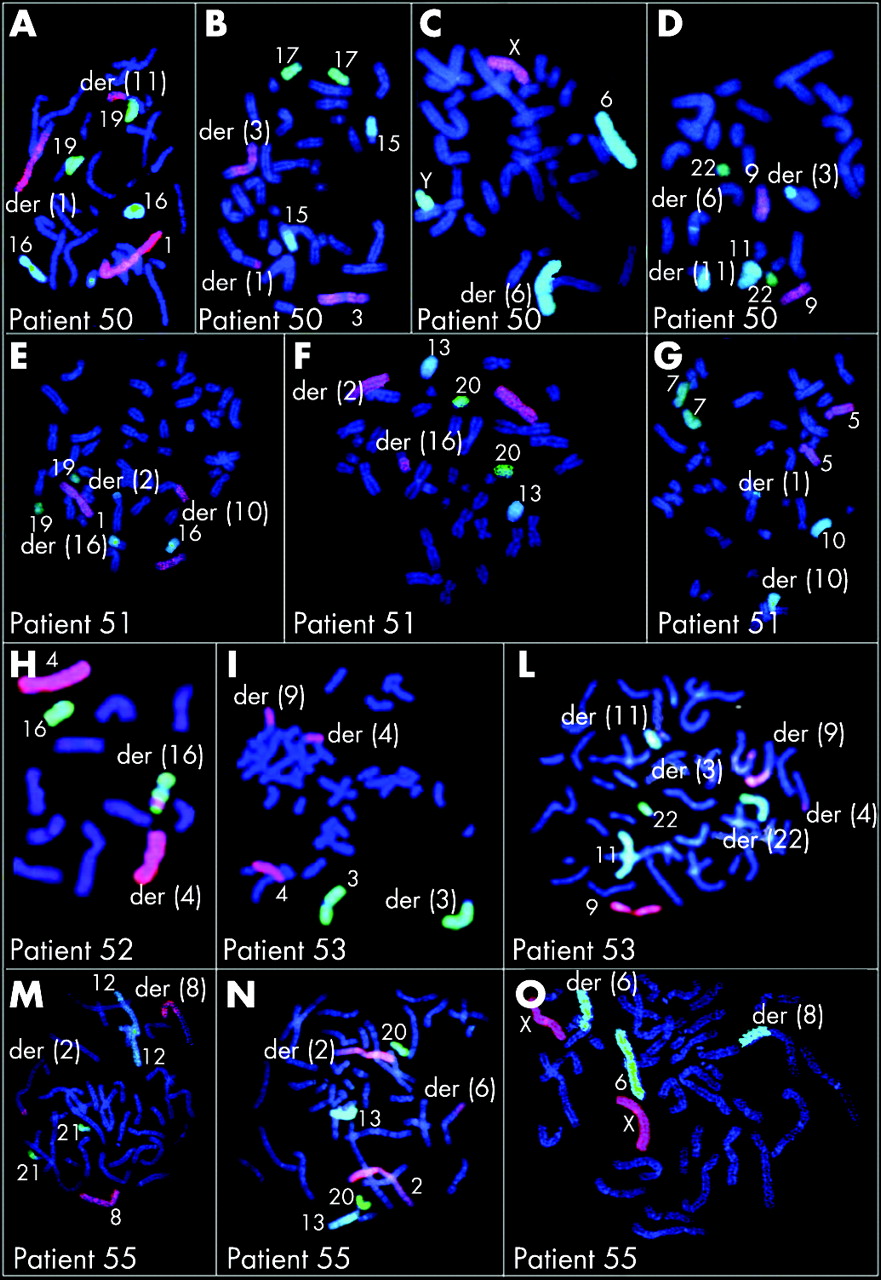
Table 7 summarises the results of array CGH in 16 patients with imbalanced CCRs; figure 2 shows array CGH profiles in patients 42–53, 55, 57, 58 and 59. The parental origin of the deletions was investigated in 11 patients (patients 42, 43, 45–51, 53, 58and was found to be paternal in all (supplementary table 2; available at http://jmg.bmj.com/supplemental). To better define the CCRs, either chromosome painting or FISH with subtelomeric probes was performed for all patients except case 57, who was investigated by SKY (figs 3–5). Chromosome painting of DNA from patients 54 and 49 has been previously reported.15 16 Please note that interpretation of breakpoints was made using the information coming both from the FISH analysis and from chromosome banding and array CGH. In most of the patients, the number of breakpoints estimated by conventional and molecular cytogenetics increased after array CGH investigations (table 7, first column). This increase was particularly evident in patients 52, 53 and 58, for whom the number of breakpoints doubled.


Array and FISH results in subjects with abnormal phenotype
Of the patients with an abnormal phenotype (42–54), all except patient 54 were unbalanced. A single deletion was found in six patients (in patients 42, 43, 44 and 48 at one of the breakpoints and in patients 45 and 46, on chromosomes or on chromosome regions unrelated to the rearrangement). Two deletions were present in patients 47, 49, 50 and 51 (six deletions at the breakpoints and two elsewhere). Three deletions were present in case 52, with one deletion at one of the breakpoints and two deletions elsewhere. Four deletions were present in case 53, two at one of the breakpoints and two elsewhere.
FISH analysis was performed with clones flanking the three deletion breakpoints of two patients (43 and 50) and confirmed that the deletions coincide with the breakpoints (fig 6A,B). In patient 43, BAC RP11–291L15 (green) mapped to a deleted region on chromosome 4 (its signal was present only on the homologous region not involved in the rearrangement) whereas RP11–6L6 (red), mapping distally to RP11–291L15, gave hybridisation signals both on the homologous region not involved in the rearrangement and on der(11). This finding demonstrated that the deletion breakpoint coincided with that of the translocation t(4;11). In patient 50, the deletion breakpoints of chromosome 6 coincided with the insertion breakpoints of chromosome 11, as RP11–93P6, mapping just distal to the last deleted probe detected by array CGH (table 7), gave FISH signals much futher from the centromere than in the normal chromosome 6, due to the insertion of 11q13–q14.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Array results in subjects with repeated spontaneous abortion and in prenatal cases
In the two women with repeated spontaneious abortion (patients 55 and 56), one (patient 55) was unbalanced with a single deletion at one of the breakpoints. All the prenatal cases (patients 57–59) were unbalanced, with a single deletion at one of the breakpoints in patient 57 and four deletions in patient 58 (all at the breakpoints), and a single deletion in a chromosome unrelated to the rearrangement in patient 59.
Breakpoints analysis
To learn more about the mechanisms by which these rearrangements originated, we investigated the surrounding sequences of each breakpoint. We searched for the presence of segmental duplications flanking the rearrangement breakpoints as described in Methods; no segmental duplications were detected. To confirm whether any common sequence motifs might have underlined these rearrangements, we narrowed down the breakpoints to a few hundred base pairs in seven patients with imbalances, using customised arrays (supplementary table 3; available at http://jmg.bmj.com/supplemental).
We compared the breakpoints sequence with the reference sequences (UCSC; http://genome.ucsc.edu/) and no particular DNA motifs (non-B DNA structures) predisposing to chromosome instability were detected.
DISCUSSION
Among the 59 patients with reciprocal translocations and CCRs, 42 deletions were found in 27 different samples. The size of the deletions ranged from 0.57 to 35 Mb, with 40 deletions being <10 Mb. The finding that the largest deletions were not identified by conventional cytogenetics is not surprising, considering the complexity of some rearrangements.
Reciprocal translocations
Warburton et al2 collected data on the frequency and outcome of apparently balanced reciprocal translocations ascertained at prenatal diagnosis and estimated that at least 6.1% is associated with serious congenital anomalies. We found that 40% (11 of 27) of patients had at least one deletion, either at one of the breakpoints or elsewhere, suggesting that the phenotypic effect is possibly due to the gene deletions, rather than the breakage of a gene. Thus, genome-wide arrays should be used for patients with a de novo translocation and an abnormal phenotype. From these findings, we might assume that in about 60% of reciprocal translocations associated with abnormal phenotypes, the breakage or the abnormal expression of dosage-sensitive genes cause the pathological phenotype. However, we cannot exclude that genome-wide arrays at higher resolution might reveal an increased percentage of pathogenic deletions in these patients. The size of the deletions varied from 0.6 Mb up to about 9 Mb. Surprisingly, deletions were detected not only at the translocation breakpoints, but also elsewhere in the genome (patients 5–7, 9). Hence, apparently balanced translocations often hide complex chromosomal rearrangements. Similar findings have been reported by Gribble et al10 and Ciccone et al.12
Complex chromosome rearrangements
Our findings show that deletions are the main cause of phenotypic abnormalities, as we found at least a deletion in 12 out 13 of these patients. Similar findings have also been reported in several papers describing single patients.11 19 20 The total size of the deletions among 11 patients with phenotypic anomalies (patients 42–47, and 49–53) varies between 0.9 and about 10 Mb.
A 35 Mb deletion was detected in a female patient who died a few hours after birth (patient 48). The complexity of the rearrangement and the poor prenatal chromosome banding made it difficult to determine if the rearrangement was really balanced. At birth, FISH analysis of the Wolf–Hischhorn critical region probe (chromosome 4p was involved in the rearrangement) showed the presence of the deletion.
However, in our series in general, there was no association between the severity of the pathology and either the number of deletions or their sizes. We could not detect any deletion in one patient with a CCR (patient 54) and profound mental retardation and congenital anomalies, although he was also tested with the 244 K platform. We also detected a cryptic deletion of about 4 Mb at one of the breakpoints of the rearrangement in one of the two normal women (patient 55) investigated for repeated spontaneous abortion.
Of the seven genes within the deletion region, COL12A1 at least is dosage-sensitive. Although no specific clinical investigations were performed, the probands did not show any connective tissue disorder. The total size of the deletions identified in the three fetuses were 7, 19 and 2 Mb, respectively. We must stress that in all patients ultrasonographic evaluations (cranial and abdominal ultrasonography and Doppler echocardiography) gave normal results, although in patient 58 clinical investigations of the terminated fetus revealed severe micrognathia.
Patient 59 showed at birth low-set abnormal ears and facial dysmorphisms. Our findings indicate that in prenatal cases with apparently balanced rearrangements, array CGH should be performed to determine whether chromosomal imbalances are present and to allow proper prognosis. Altogether, the number of deletions does not seem to be strictly related to the number of breakpoints: patient 57, with 15 breakpoints, has a single deletion, whereas patient 52, with six breakpoints, has three deletions. This contradicts the idea that there would be a relationship between the number of chromosome breaks and the extent of the phenotypic effect.7
Mechanisms of formation of rearrangements having cryptic imbalances
Reciprocal translocations
We tested the parental origin in five patients and found it to be exclusively paternal. This is in agreement with the findings in all de novo reciprocal translocations patients with cryptic imbalances reported by Gribble et al10 (three patients) and by Ciccone et al12 (one patient; patient 3). Thus paternal meiosis seems to be more prone to chromosomal exchange, although it is not clear if this preference applies only to translocations associated with deletions, both at the translocation breakpoints and elsewhere, or if it simply reflects a tendency of male meiosis to form translocations. The finding that the majority of interstitial deletions is paternal in origin21 might indicate that the mechanisms leading to deletions are more frequent during male meiosis, although the situation is far from clear. Several studies have focused on the most common non-robertsonian translocation, the t(11;22)(q23;q11), and found this rearrangement in DNA from sperm but not in somatic tissues isolated from karyotypically normal individuals, indicating that de novo translocations do occur during male meiosis. However it is not known whether translocation also arises in female meiosis.22 It is clear that this situation is completely different from robertsonian translocations, in which a marked preference for maternal origin has been demonstrated. The same finding has been shown for all the recurrent de novo der(4)t(4;8)(p16;p23) tested for parental origin,23 which were found to occur during maternal meiosis. Different mechanisms of formation for robertsonian translocations and the two most common translocations, t(11;22) and t(4;8)(p16;p23), have been hypothesised. Analysis of many unrelated patients with t(11;22)(q23;q11) found that breakpoints occur within palindromic AT-rich repeats (PATRRs) on 11q23 and 22q11 (PATRR11 and PATRR22). The majority of the breakpoints are localised at the centre of the PATRRs, suggesting that the center of the palindrome is susceptible to double-strand breaks (DSBs), thereby inducing incorrect chromosomal rearrangement.24 Concerning robertsonian translocations, although the mechanism of formation is unknown, these translocations may originate during oogenesis due to a double-strand break (DSB) initiated by the genomic architectural features and/or DNA sequence at the short arm region of acrocentric chromosomes. Meiotic pairing between non-homologous chromosomes and the aberrant repair of the DSBs by recombination between homologous sequences shared between these acrocentric chromosomes, may lead to the formation of robertsonian translocations.25 A different mechanism has been suggested to be responsible for the recurrent translocation t(4;8)(p16;p23), the breakpoints of which coincide with homologous segmental duplications23 favouring the occurrence of non-allelic homologous crossing-over between non-homologous chromosomes. We could not find segmental duplications at the breakpoints in any of the translocations we screened by array CGH. The genome-wide array CGH definition of the deletion breakpoints is too large both to identify possible non-B DNA structures leading to the translocation as reported for t(11;22) and to discern if there are special DNA sequences that predispose to the occurrence of a deletion at the breakpoint. For those patients in whom the imbalance involves chromosomes or chromosomal regions different from the translocated regions, we have to assume the same mechanisms lead to CCRS.
Complex rearrangements
We investigated 15deletions found in complex rearrangements for the parental origin and all turned out to be paternal. In patients 47, 50, 51 and 53, paternity was confirmed using microsatellite markers for both deletions present in each patient, but in patient 58 for only one of the four deletions. Therefore, the aforementioned propensity of male germ cells to generate reciprocal translocations associated with cryptic deletion is further reinforced by the findings that all 11/16 CCRs associated with deletion that were analysed for the parental origin were found to be paternal. As constitutional chromosome abnormalities such as translocations or marker chromosomes may cause male infertility with arrest very early in meiosis I,26–29 it seems likely that the CCRs occurred at this point and not during the previous spermatogonial proliferation, when the mechanisms for recognising and correcting or eliminating cells with errors would have blocked the cells as soon as meiosis began. Confirming this, CCRs are rarely transmitted through spermatogenesis and are frequently ascertained by male infertility.8
It is important to stress that none of the deletion or translocation breakpoints of our CCRs coincided with genomic regions known to be associated with instability such as segmental duplications.30 Moreover, the narrowing of 22 breakpoints to a few hundred base pairs in 7 patients and the comparison of the deleted regions with the reference genome (UCSC) excluded their coincidence with a non-B DNA structure known to have a potential for genomic instability.31–33 These findings indicate that the basic mechanism responsible for occurrence of CCRs is not linked to particular DNA structures and/or sequences, but to some, possibly external, factors acting at meiosis I.
We cannot exclude that CCRs are the consequence of an improper repair of the DSBs that are the initiating event in normal recombination.34 A similar situation with activation of the recombination pathway not resulting in functional exchanges has been reported in an azoospermic man with meiosis I arrest.35 The fathers of our patients are apparently fully fertile; except for two families with only a single child, all families have multiple children and no problem in conceiving has been reported. Moreover, only one of the fathers had any history of cancer that might have suggested an intrinsic genomic instability;36 this man, the father of patient 51, had developed Hodgkin’s lymphoma 8 years before the child’s conception. Regardless of the underlying cause of formation of these abnormal sperm, we have to assume that it is, at least predominantly, linked to spermatogenesis. This is in contrast with the higher female vulnerability to non-disjunction in meiosis I that led Hunt and Hassold37 to hypothesise that either more errors occur during oogenesis, or that the mechanisms for recognising and correcting or eliminating cells with errors are more efficient in spermatogenesis.
CONCLUSION
This study shows that:
(1) Phenotypic abnormalities, present in subjects with “balanced” chromosome rearrangements occur in 40% of translocation patients and in most patients with CCRs, due to small deletions, most of which (27 of 42) occur the breakpoints. The lack of association between the size and number of deletions and the severity of the phenotype obviously depends on the gene content of the unbalanced regions and the genomic background.
(2) All the deletions we analysed fully for the parental origin (5 patients with a reciprocal translocation and 11 patients with CCRs) originated during paternal meiosis. This suggests that spermatogenesis is a very delicate moment susceptible to a type of “explosion” of chromosomes that determines either reciprocal translocations or CCRs. We could not find specific DNA sequences at the 22 breakpoints identified using a specific customised array. Thus, we were unable to determine which mechanisms are behind the concurrent breakage of several chromosomes with loss of part of the broken portions and random assortment. Considering that all the men who fathered children with unbalanced translocations or CCRs are fertile, we can hypothesise that during spermatogenesis some cells escape the mechanism controlling correct crossing-over, undergoing chaotic break and reunion of several chromosomes and exposing the broken portions to exonuclease degradation.
(3) Several patients interpreted as having simple reciprocal translocations in fact had CCRs with >3 breakpoints (5 of 27).
To conclude, once again array CGH allowed new insights into cytogenetic diagnosis and research, showing that deletions may be common in apparently balanced rearrangements associated with abnormal phenotypes.
Supplementary material is available on the JMG website at http://jmg.bmj.com/supplemental
Acknowledgments
This work was supported by the Telethon foundation (to T Pramparo GGP05177) and PRIN 2005, Fondazione Mariani and Fondazione CARIPLO (all to O Zuffardi). MR and AS received a grant from the Swiss National Foundation (No. 32-113635/1).
REFERENCES
Supplementary materials
web only tables 44/12/750
Files in this Data Supplement:
Footnotes
Competing interests: none declared.
- Abbreviations:
- BAC
- bacterial artificial chromosome
- CCR
- complex chromosome rearrangement
- CGH
- comparative genome hybridisation
- DMD
- Duchenne muscular dystrophy
- DSB
- double-strand break
- FISH
- flurorescence in situ hybridisation
- PATRR
- aplindromic AT-rich repeat
- STR
- short tandem repeat