Article Text

Abstract
Background Nephrolithiasis (NL) is a complex multifactorial disease affecting up to 10%–20% of the human population and causing a significant burden on public health systems worldwide. It results from a combination of environmental and genetic factors. Hyperoxaluria is a major risk factor for NL.
Methods We used a whole exome-based approach in a patient with calcium oxalate NL. The effects of the mutation were characterised using cell culture and in silico analyses.
Results We identified a rare heterozygous missense mutation (c.1519C>T/p.R507W) in the SLC26A6 gene that encodes a secretory oxalate transporter. This mutation cosegregated with hyperoxaluria in the family. In vitro characterisation of mutant SLC26A6 demonstrated that Cl−-dependent oxalate transport was dramatically reduced because the mutation affects both SLC26A6 transport activity and membrane surface expression. Cotransfection studies demonstrated strong dominant-negative effects of the mutant on the wild-type protein indicating that the phenotype of patients heterozygous for this mutation may be more severe than predicted by haploinsufficiency alone.
Conclusion Our study is in line with previous observations made in the mouse showing that SLC26A6 inactivation can cause inherited enteric hyperoxaluria with calcium oxalate NL. Consistent with an enteric form of hyperoxaluria, we observed a beneficial effect of increasing calcium in the patient’s diet to reduce urinary oxalate excretion.
- urology
- nephrology
- mutation
- missense
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Introduction
Nephrolithiasis (NL) is the second most frequent renal disease after hypertension affecting up to 10% of individuals worldwide, and in nearly all countries, its prevalence and incidence are reported to be rising.1 NL is highly recurrent and is a major cause of patient pain, disability and lost working days. It is also associated with increased risks of cardiovascular disease, bone demineralisation and fractures, chronic kidney disease and end-stage renal disease.2–4
The vast majority of kidney stones is composed of calcium oxalate.5 Even a small increase in urinary oxalate markedly promotes calcium oxalate crystal formation,6–8 and hence, hyperoxaluria is a major risk factor for kidney stones.7 The oxalate found in urine is either from exogenous or endogenous origin. Dietary oxalate, which accounts for almost 50% of the oxalate excreted in urine,9 is abundantly present in plant and animal sources, absorbed by the gut and excreted unchanged in the urine.9 In addition, endogenous oxalate production occurs in the liver resulting from the metabolism of glyoxylate.10 11 Primary hyperoxaluria (PH) is an inherited autosomal recessive metabolic disorder that results from increased endogenous oxalate production, whereas secondary hyperoxaluria is acquired and caused by excessive intake of dietary oxalate or of oxalate precursors or by factors that can increase net oxalate absorption by the gastrointestinal tract (eg, low calcium diet, lipid malabsorption and abnormal microbiome). Therefore, identification of the potential mechanisms leading to hyperoxaluria is an important step to optimise care and treatment of oxalate-dependent NL.
In mice, ablation of the oxalate transporter SLC26A6 causes a defect in intestinal oxalate secretion that results in increased net absorption of dietary oxalate,12 leading to hyperoxaluria and calcium oxalate NL.12 13 This observation supports the possibility that an additional type of inherited hyperoxaluria might exist in human patients due to mutations leading to defective intestinal oxalate secretion, thereby resulting in enhanced net intestinal absorption of oxalate. Four studies have assessed the impact of SLC26A6 variants in humans.14–17 However, these studies either reported no association of SLC26A6 variants with hyperoxaluria or with NL,14–16 or reported mutations that disrupt a complex between SLC26A6 and the citrate transporter NaDC-1 thereby causing hypocitraturia as a risk factor for NL.17 Therefore, no rare human SLC26A6 variant contributing to hyperoxaluria has been reported so far.
Here, we report a family in which a rare heterozygous missense mutation in the SLC26A6 gene is associated with hyperoxaluria and recurrent bilateral calcium oxalate NL. In vitro characterisation of the mutant demonstrated that the mutation leads to defective membrane expression and oxalate transport activity with evidence of a dominant negative effect on wild-type (WT) transporter. We conclude that rare, deleterious mutations in SLC26A6 gene are a possible cause of inherited hyperoxaluria and therefore can promote NL in humans.
Concise methods
A detailed description of material and methods is available as online supplemental information.
Supplemental material
Patients testing
Biochemical and hormone measurements were performed using standard techniques as detailed in online supplemental materials.
Participants from UK Biobank
We analysed up to 200 619 samples with available exome sequencing data. This research is part of UK Biobank research application #67 575. The strategy used in this study to identify patients with kidney stones is reported in online supplemental materials.
DNA sequence analyses
The DNA proband was assessed through whole-exome sequencing at the genotyping platform of the European Genomic Institute for Diabetes (Lille, France). For this purpose, we used the Human Core Exome EF Multiplex Complete kit (Twist Bioscience) in combination with Illumina next-generation sequencing.
The SLC26A6 mutation was confirmed in the proband through Sanger sequencing, and then assessed in the family members. Primer sequences and PCR conditions are available on request. Fragments were sequenced in both directions and subsequently analysed using the 3730xl DNA Analyzer (Applied Biosystems). Electropherogram reads were assembled and examined using the SeqScape software (Applied Biosystems).
Detection of variants in UK Biobank
We used exome data from pVCF format (field #23 156). Only variants with a coverage higher than 10 reads and quality Genotype quality (GQ) score higher than 20 were kept for further analyses. Annotation of variants was done using the Ensembl Variant Effect Predictor tool V.103 (RefSeq).
In silico analyses of the effects of the p.R507W mutation on Slc26a6 protein
A structural model of human SLC26A6 was developed using the experimental structure of the mouse SLC26A9 homodimer anion transporter as structural template.18 Different structural bioinformatics approaches were used to investigate the model structure and the amino acid replacement including the computations of ΔΔG values between the WT and the variant proteins. The different approaches used and additional structural analyses are reported in the online supplemental material section.
SLC26A6 expression constructs
An HA-tagged wild-type human SLC26A6 cDNA expression construct (HA-WT)19 was used to generate HA-tagged and myc-tagged p.R507W human SLC26A6 mutants (HA-MT and myc-MT, respectively) with a Q5 site-directed mutagenesis kit (New England Biolabs).
Cell culture and transfections
OKP cells (American Type Culture Collection(ATTC)) were used for all transfection experiments. 1×105 OKP cells per well of a 24 wells cell culture dish were reverse transiently transfected with 2 µL of Lipofectamine 2000 (Thermo Scientific) and 0.1–0.25 µg of HA-WT, myc-WT or HA-MT cDNA as indicated in the respective figure legend for each experiment. Cells were assayed 72 hours post-transfection.
Cell surface biotinylation
OKP cells were surface biotinylated 72 hours post-transfection as described previously.19 Membranes were probed with a rabbit anti-SLC26A6 polyclonal antibody19 (R29; 1:50 000 dilution), a rabbit anti-HA polyclonal antibody (71–5500; 1:250 dilution; Thermo Scientific) or a mouse anti-myc monoclonal antibody (R950-25; 1:5000 dilution; Thermo Scientific). Primary antibody labelling was detected with either a donkey antirabbit or a donkey antimouse Horse Radish Peroxydase (HRP-labelled secondary antibody (711-035-152 and 715-035-150 respectively; 1:20 000 dilution; Jackson ImmunoResearch),19 visualised by enhanced chemiluminescence (Clarity Western ECL Substrate; Bio-Rad) and recorded on film.
Coimmunoprecipitation
Transfected OKP cells were cultured for 72 hours prior to solubilisation with 1% digitonin. myc-tagged WT SLC26A6 was immunoprecipitated from OKP cell detergent lysates with the anti-myc antibody R950-25 (Thermo; 5 µg IgG/mL lysate). Immunoprecipitates were captured with Protein G Sepharose Fast Flow (Sigma P3296), and then Sepharose beads were washed extensively with a series of normal and high salt washes to reduce non-specific binding. Immunoprecipitates were released from sepharose beads by incubation with 2× Sodium Dodecyl Sulfate (SDS)-PAGE sample buffer containing 100 mM dithiothreitol (DTT) for 2 min at 100°C. The immunoprecipitates were subjected to SDS-PAGE, transferred to a polyvinylidene fluoride (PVDF membrane and then analysed by western blot with antibodies directed against the HA-epitope and myc-epitope tags (71–5500 and R950-25 antibodies, respectively).
Cl−-dependent 14C-oxalate uptake
See reference ref 19 for a detailed description of the rationale and methodology behind the assessment of Cl−-gradient dependent 14C-oxalate uptake mediated by the SLC26A6 expression constructs (HA-WT and HA-MT) when transfected into OKP cells. Briefly, uptakes were performed for each transfection condition in the presence and absence of an outwardly directed chloride gradient to provide an estimate of the proportion of the total 14C-oxalate cell uptake that was specifically chloride dependent. Uptakes were also performed in untransfected cells (exposed to transfection reagent alone) to provide an estimate of background levels of endogenous chloride-dependent 14C-oxalate uptake. The background values were then subtracted from the values in transfected cells to determine the level of chloride-dependent 14C-oxalate uptake that is directly and solely attributable to the activity of the transfected SLC26A6 expression constructs.
Statistics
Data values are presented as mean±SEM. Statistical significance was evaluated by unpaired two-tailed Student’s t-test (GraphPad Prism).
Results
Primary clinical data of the proband
The patient is a late adolescent woman from non-consanguineous parents. She has a personal history of Hashimoto’s thyroiditis diagnosed during her early adolescence and replacement with L-thyroxine. She had her first renal colic during her early adolescence. Since then, she has had recurrent renal colic (>20) and passed multiple kidney stones. Abdominal CT and ultrasound detected multiple and bilateral stones without nephrocalcinosis (online supplemental figure S1). A spontaneously evacuated kidney stone was analysed by infrared spectrometry revealing a composition of calcium oxalate monohydrate (~80% whewellite) suggesting that NL is oxalate dependent. Biochemical analyses, performed in the absence of any treatment and 2 months after her last renal colic (table 1), identified the presence of frank hyperoxaluria (733 µmol/day, N: 40–330), which was confirmed on a second independent urine sampling (800 µmol/day, N: 40–330). Adequacy of urine collection was attested by measuring the daily excretion of creatinine (0.16 mmol/kg/day, N: 0.12–0.19). Interview with a dietitian excluded excessive intake in oxalate-rich aliments (~50 mg/day) and revealed very low calcium intake (~400 mg/day). The patient had no history of bowel disease or diarrhoea and showed no sign of malabsorption. Table 1 also showed no biological evidence of malabsorption. Kidney stone-causing conditions like vitamin D excess or primary systemic disease (eg, primary hyperparathyroidism or renal leak of phosphate) were excluded. The calcium-loading test detected increased digestive absorption of calcium as evidenced by the increased delta between postcalcium load calciuria and fasting calciuria. However, the patient did not exhibit hypercalciuria. The only other risk factor for NL was very mild hypocitraturia (1.18 mmol/24 hours, N>1.67). The patient exhibited no extrarenal symptoms or biological abnormalities suggesting systemic oxalosis, and renal function appeared normal.
Supplemental material

In silico analysis of the putative effects of the R507W substitution on the SLC26A6 protein structure. The two subunits are presented with one subunit coloured in green and the other in orange (cartoon representation). The olive spheres represent the boundary of the lipid bilayer as computed with the PPM server. A zone predicted to interact with the Cl− ion as proposed in the mouse SLC26A9 experimental structure is highlighted by a black dashed circle in subunit a for orientation. R507 residues of both subunits are flagged and coloured in blue; this residue is located on the last helix of the transmembrane domain 22 residues prior to the beginning of the Sulfate Transporter Antagonist of anti-Sigma factor (STAS domain. The insertion loop that could not be built is shown as a dashed line. The mutation is expected to destabilise the protein and to impede optimal insertion into the lipid bilayer.
Characteristics, blood and urine analyses and calcium-loading test of the patients
Identification of a rare heterozygous missense mutation in Slc26a6
Through whole-exome sequencing of the proband’s DNA, we investigated rare coding variants of potential interest with minor allele frequency below 0.1% in the Genome Aggregation Database (GnomAD V.2.1.1) in a list of genes or candidate genes linked with NL (online supplemental table 1). We did not find any pathogenic or likely pathogenic variant according to the criteria of the American College of Medical Genetics and Genomics.20 No pathogenic or likely pathogenic variant or variant of unknown significance was detected in the three genes involved in PH. However, we found a rare heterozygous variant of unknown significance located in the 13th coding exon of SLC26A6 (NM_022911.2: c.1519C>T; p.R507W). SLC26A6 encodes an oxalate transporter involved in hyperoxaluria and NL based on mouse knockout studies.12 13 According to GnomAD (V.2.1.1), SLC26A6 is intolerant to loss-of-function variation with an observed/expected score of 0.65 (0.48–0.91), and the p.R507W mutation is very rare, as it has been found in only 5 out of 280 674 alleles reported in GnomAD (in Europeans and South Asians). According to SIFT, Mutation Taster and PolyPhen-2 (HumDiv), the p.R507W mutation was predicted to be deleterious, disease causing and damaging, respectively.21–23
Supplemental material
Human SLC26A6 residue p.R507 was found fully conserved in an interspecies comparison. The conservation of this residue was also very high in this family of proteins (ie, the score at position 507 was equal to 8, the maximum is 9, via the Consurf server). We then built a homology model using as experimental template the mouse Slc26a9 homodimer anion transporter tri-dimensional (3D structure (figure 1). The modelling was possible as the overall sequence identity between the two proteins is around 40% (see online supplemental materials).
The human SLC26A6 model structure positioned into a membrane is shown in figure 1. p.R507 is located on the C-terminal side of the last transmembrane helix, prior to the STAS domain. The region of p.R507 is predicted to be essentially rigid (PredyFlexy computation) and thus not very tolerant to the p.R507W substitution, which was predicted to likely perturb correct interaction with the membrane. The details of our in silico analyses are provided as online supplemental materials.
Family investigation of the Slc26a6 p.R507W mutation
Since the SLC26A6 mutation was potentially pathogenic, both parents of the proband were assessed. Sanger sequencing identified the same heterozygous SLC26A6 mutation in the father, while the mother was WT (online supplemental figure S2). The mother had no personal or familial history of kidney stones and exhibited normal levels of urinary oxalate (250 µmol/day, N: 40–330).

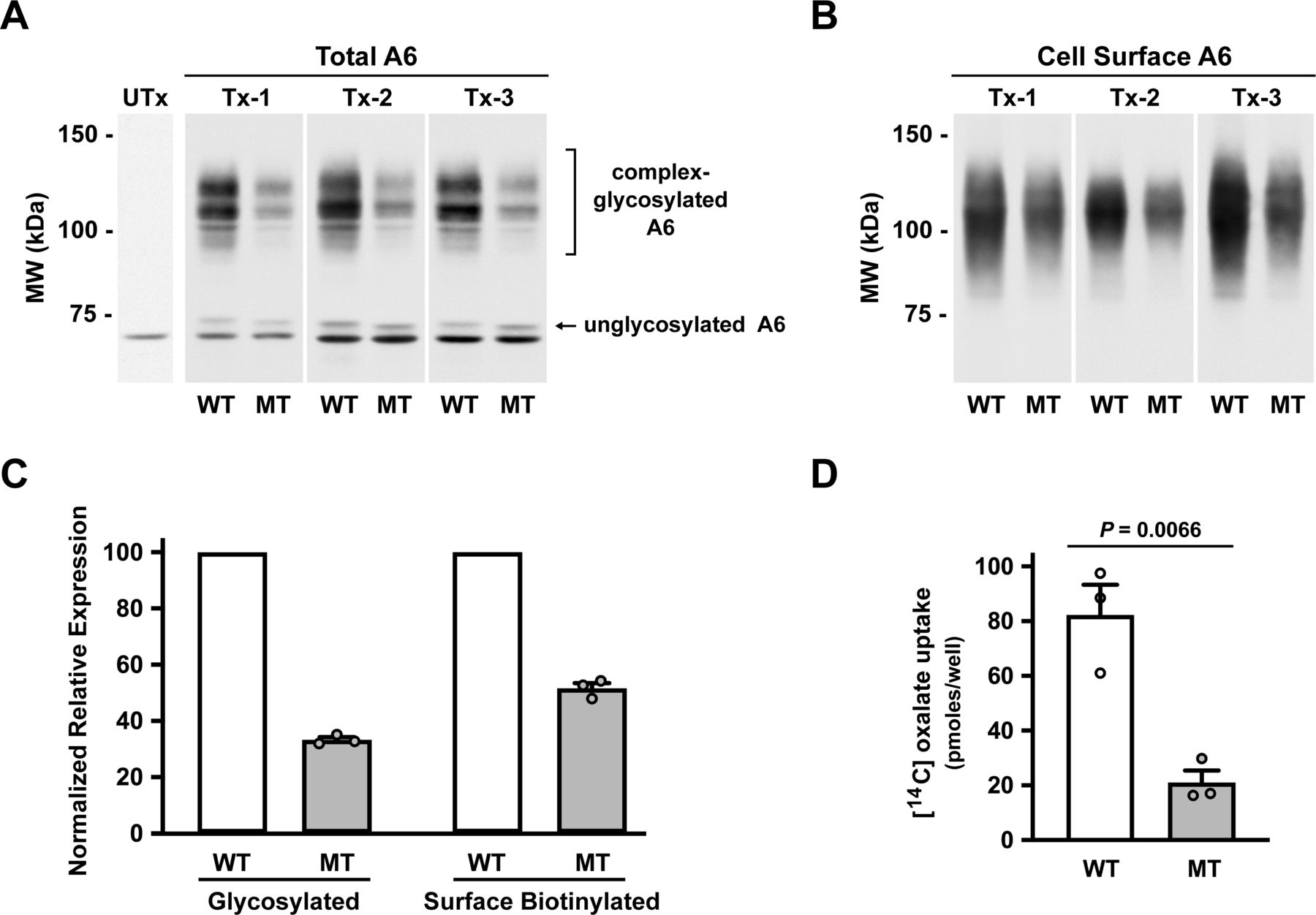
Characterisation of the R507W SLC26A6 mutation in transfected OKP cells. OKP cells were transfected with equivalent amounts (0.1 µg cDNA per well of a 24-well dish) of either wild-type (WT; HA-WT) or mutant (MT; HA-MT) human SLC26A6 cDNA. Cells were assayed 72 hours post-transfection. For each transfection (Tx-1 to 3), uptakes and companion biotinylations were performed in triplicate. Utx, untransfected control. Panels A+B show representative images selected from each transfection. Western blots were probed with the antihuman Slc26a6 polyclonal antibody, R29. Panel A: western analysis of total cell lysates isolated from each transfection condition. Panel B: western analysis of cell surface biotinylatable hA6 from each transfection condition. Panel C: expression levels of total and cell surface biotinylatable hA6. Expression levels were determined by densitometry. To facilitate comparisons between transfections (n=3 independent transfections), MT expression levels for each transfection were normalised to mean densitometry values for the WT densitometry values for each transfection. All WT levels are expressed as 100 (%), and the individual MT values vary accordingly. Panel D: chloride-dependent 14C-oxalate uptake attributable to transfected wild-type (WT; HA-WT) and mutant (MT; HA-MT) Slc26a6. The uptake values are presented as pmoles 14C-oxalate uptake per well of a 24-well plate and are not normalised to levels of expressed protein. WT, wild type; HA-WT, hemagglutinin-tagged wild type; MT, mutant; HA-MT, hemagglutinin-tagged mutant; tx, transfection; Utx, untransfected.
Even though the father did not report any renal colic, biochemical analyses revealed that he also had hyperoxaluria (520 µmol/day, N: 40–330). The calcium loading test was strictly normal, and no other risk factor for NL was identified for the father (table 1). However, we noticed that he exhibited a urine output of almost 4 L/day reflecting a very high-water intake. This high urine output may explain why he never experienced kidney stones. Furthermore, contrasting with the proband, he did not exhibit any hypocitraturia. No other relatives from same family were accessible for medical examination and genetic testing.
Response to medical treatment
We hypothesised that the proband presented with inherited enteric hyperoxaluria due to deficient SLC26A6-mediated oxalate secretion resulting in increased net absorption of dietary oxalate. We advised her to increase drinking water intake in order to reach a urinary output of at least 3 L/day and to systematically add a dairy product to each meal in order to increase her calcium intake up to the recommended daily amounts of 900 mg/day. The latter intervention turned out to be very effective, as urinary oxalate excretion markedly dropped from >700 µmol/day down to ~300 µmol/day on repeated control analyses while the mild hypocitraturia persisted (1.49 mmol/24 hours N<1.67). No other medication was initiated, and NL also dramatically improved, as she has not experienced any recurrence of kidney stones for 2 years of follow-up.
Physiological characterisation of the variant
In order to assess the pathogenicity of the SLC26A6 p.R507W mutation, we next assessed its effect on protein expression, protein trafficking and chloride-dependent oxalate transport. Under physiological conditions, apical membrane SLC26A6 in epithelial cells mediates oxalate secretion by operating in the direction of exchanging intracellular oxalate for extracellular chloride. However, SLC26A6 can mediate Cl−-oxalate exchange in either direction depending on the direction of the net driving force. We measured Cl−-gradient stimulated 14C-oxalate uptake as a measure of oxalate transport activity mediated by human SLC26A6 transfected into OKP epithelial cells, as in previous studies of SLC26A6 glycosylation mutants.19 We specifically evaluated the chloride-dependent component of 14C-oxalate uptake that was mediated by the transfected human SLC26A6 expression constructs by correcting for endogenous oxalate transport (see Methods and online supplemental figure S3 for details).

The R507W mutation directly impairs chloride-dependent oxalate transport capability in human SLC26A6. The initial characterisation of the Δ507 mutation suggested that the degree of inhibition of the Slc26a6-mediated chloride-dependent oxalate transport activity was significantly greater than that predicted by decreased protein expression alone. To better address that possibility, we transfected OKP cells with 2.5× more mutant (MT) than wild-type (WT) human SLC26A6 (hA6) in an effort to significantly enhance the apparent transport activity of Δ507-hA6. Cells were transfected with either 0.25 µg of HA-MT (MT) or 0.1 µg HA-WT (WT) cDNA per well of a 24-well plate. The experiments were performed and data presented identically as in panels A–C of figure 1. Panel A: western analysis of total cell lysates isolated from each transfection condition. Panel B: western analysis of cell surface biotinylatable hA6 from each transfection condition. Panel C: expression levels of total and cell surface biotinylatable hA6. Expression levels were determined by densitometry. All WT levels are expressed as 100 (%), and the individual MT values vary accordingly. Panel D: chloride-dependent 14C-oxalate uptake attributable to transfected wild-type (WT; HA-WT) and mutant (MT; HA-MT) Slc26a6. The principal objective of this experiment was to directly address the transport capability of mutant relative to wild-type Slc26a6 independent of protein expression. To that end, the transport data presented in panel D were normalised to cell surface biotinylatable hA6 for each transfection. tx, transfection; Utx, untransfected.
Characterisation of the effect of the p.R507W mutation on expression and function of human SLC26A6 transfected into OKP cells is shown in figure 2. Results are illustrated for three independent experiments in which OKP cells were transfected with identical amounts of cDNA encoding the WT and mutant (MT) transporters. Figure 2A shows immunoblots of total cell lysate and compared transfected cells with an untransfected control lane. Transfection of OKP cells results in expression of a minor band just under 75 kDa and multiple prominent bands >100 kDa. As demonstrated in a previous study, the lower band corresponds in size to nascent unglycosylated SLC26A6, whereas the upper bands correspond to complex-glycosylated SLC26A6.19 Surface biotinylated SLC26A6 is shown in figure 2B, corresponding in size to the complex-glycosylated forms of SLC26A6. As illustrated by the immunoblots in figure 2A and B and summarised by the densitometry in figure 2C, there was a marked reduction in glycosylated and cell surface expression of the mutant transporter compared with WT. Although these findings indicate that the p.R507W mutation leads to decreased processing and trafficking, and/or increased degradation of the transporter, the qualitative similarity of the western blot molecular weight band profiles of the WT and MT constructs strongly suggests that the mutation does not directly affect the glycosylation of SLC26A6 per se. As shown in figure 2D, chloride-dependent 14C-oxalate uptake mediated by mutant SLC26A6 was also markedly impaired. Of interest, whereas the mutation resulted in approximately a 50% reduction in cell surface expression of SLC26A6 (figure 2C), it led to a disproportionately higher reduction of Cl−-dependent oxalate transport activity of approximately 75%. This suggested that the mutation might affect both the expression and the intrinsic transport capability of SLC26A6.
We therefore next designed an experiment to test directly whether the mutation causes a defect in the intrinsic transport function of SLC26A6 in addition to the defect in net surface expression demonstrated in figure 2. Cells were transfected with 2.5-fold more mutant than WT cDNA with the goal of approximately equalising surface expression of the mutant and WT transporters. As illustrated by the immunoblots in figure 3A and B and summarised by the densitometry in figure 3C, the protocol achieved roughly equivalent amounts of the glycosylated and surface expressed forms of the mutant and WT transporters. Most importantly, assessment of transport activity normalised to surface expression, as shown in figure 3D, indicated that the p.R507W mutation does indeed cause a marked defect (approximately 50%) in intrinsic transport activity of SLC26A6. The findings of figures 2 and 3 taken together suggest that the 75% defect in transport activity of mutant compared with WT SLC26A6 when equal amounts of cDNA are transfected (figure 2D) results from about a 50% defect in cell surface expression (figure 2C) and a roughly 50% defect in intrinsic transporter activity (figure 3D).
Given that SLC26 family transporters exist as dimers,18 24 we conducted additional studies to test the possibility of a dominant negative effect of the p.R507W mutation on expression of the WT transporter. Our approach was to assay expression of myc-tagged WT SLC26A6 as a function of cotransfection with either HA-tagged WT or HA-tagged mutant transporter. We chose transfection conditions to achieve equivalent expression levels of HA-WT and HA-MT SLC26A6 (online supplemental figure S4). As illustrated by the immunoblots in figure 4A and B, and summarised by the densitometry in figure 4C, coexpression of mutant SLC26A6 caused 70% reduction in glycosylated and cell surface expression of myc-tagged WT SLC26A6 compared with coexpression of WT SLC26A6. These results support the possibility that the p.R507W mutation may have a dominant negative effect on expression of the WT transporter so that the hyperoxaluria phenotype of patients heterozygous for this mutation may be more severe than predicted by haploinsufficiency alone.

Coexpression of the R507W mutant significantly reduces both total and cell surface expression of wild-type SLC26A6. OKP cells were cotransfected with 0.1 µg myc-tagged human SLC26A6 (myc-WT) cDNA and either 0.1 µg HA-tagged wild-type Slc26a6 (HA-WT) cDNA or 0.25 µg HA-tagged Arg507Trp mutant Slc26a6 (HA-MT) cDNA per well of a 24-well dish. Cotransfection conditions were selected to achieve equivalent levels of expression of each HA-tagged cDNA construct to facilitate a direct comparison of the effects of coexpression of each protein. Cells were assayed 72 hours postcotransfection. See online supplemental figure S3 for a representative cotransfection experiment illustrating simultaneous expression of each epitope-tagged construct. Panel A: western analysis of myc-tagged wild-type Slc26a6 expression in total cell lysates isolated from each cotransfection. Panel B: western analysis of cell surface biotinylatable myc-tagged Slc26a6 from each cotransfection. Western blots depicted in panels A+B were probed with a mouse anti-myc monoclonal antibody (Thermo Fisher R950-25; 1:5000 dilution). Panel C: expression levels of total and cell surface biotinylatable myc-tagged Slc26a6. Expression levels were determined by densitometry. To facilitate comparisons between transfections (n=4 independent transfections) myc-WT:HA-MT cotransfection expression levels were expressed relative to the densitometry values for myc-WT:HA-WT cotransfection expression levels for each transfection and myc-WT:HA-WT cotransfection expression levels were normalised to a mean value of 100 (%). Co-IP, coimmunoprecipitation; Co-Tx, cotransfection; IP, immunoprecipitation; Tx, transfection; Utx, untransfected.
We next performed a series of WT/MT co-immunoprecipitation studies to verify the association of MT with WT transporter to explain the dominant negative phenotype. OKP cells were cotransfected with myc-tagged WT SLC26A6 and either HA-tagged WT SLC26A6 or HA-tagged MT SLC26A6 as described for the experiment outlined in figure 4. The cells were solubilised with 1% digitonin, subjected to immunoprecipitation with an anti-myc antibody, and the resulting immunoprecipitates were analysed by SDS-PAGE and western blot with antibodies directed against either the myc-tag or the HA-tag (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The R507W substitution does not inhibit association of MT-A6 with WT-A6. The principal objective of this experiment was to determine if the p.R507W mutation affected the ability of the resultant mutant to associate with wild-type (WT) Slc26a6 and form the prototypic Slc26a6 multimeric complex. By using different epitope tags for the potential binding partners, we used anti-myc tag (WT) primary immunoprecipitation to assess association with HA-tagged protomers (WT +MT) by monitoring levels of coimmunoprecipitation. OKP cells were cotransfected (Co-Tx) as described for figure 3, solubilised with 1% digitonin and subjected to immunoprecipitation with a mouse anti-myc monoclonal antibody (Thermo Fisher R950-25; 5 µg IgG/mL lysate). Panel A: western analysis of myc-tagged WT Slc26a6 expression in total cell lysates isolated from each cotransfection. Panel B: western analysis of HA-tagged Slc26a6 expression in total cell lysates isolated from each cotransfection. Panel C: western analysis of primary immunoprecipitated myc-tagged WT Slc26a6 from each cotransfection. Panel D: western analysis of coimmunoprecipitated HA-tagged Slc26a6 from each cotransfection. Western blots depicted in panels A+C were probed with anti-myc antibody R950-25 (1:5000 dilution) and those in panels B+D were probed with anti-HA antibody 71–5500 (1:50 dilution). Panel E: relative expression of primary immunoprecipitated myc-tagged WT Slc26a6 and coimmunoprecipitated HA-tagged WT or mutant Slc26a6. Expression levels were determined by densitometry. To facilitate comparisons between cotransfections (n=3 individual cotransfection events; Tx-1 to Tx-3), protein expression levels determined for the myc-WT/HA-MT cotransfection condition were expressed relative to the densitometry values for the myc-WT/HA-WT cotransfection condition for each experiment, and myc-WT:HA-WT cotransfection expression levels were normalised to 100 (%). *Not significantly different at p=0.3566. tx, transfection; Utx, untransfected.
As observed in the previous experiment (figure 4), cotransfection of mutant HA-A6 with wild-type myc-A6 led to a dramatic decrease in expression of mature WT myc-A6 in both the solubilised lysate (figure 5A) and the companion primary immunoprecipitation (figure 5C). Western analysis of the solubilised lysate with the anti-HA antibody (figure 5B) confirmed that cotransfection with 2.5× HA-MT relative to HA-WT resulted in roughly similar levels of expression of the two HA-tagged SLC26A6 expression constructs. We observed significant coimmunoprecipitation of both wild-type and mutant HA-A6 with the myc-tagged WT-A6 (figure 5D). Despite the presence of similar amounts of HA-WT and HA-MT in the initial cell lysates for each experiment (figure 5B), their relative recoveries by coimmunoprecipitation could only reflect, at best, the relative recovery of the primary immunoprecipitated binding partner (myc-WT). If the two constructs multimerise with wild-type SLC26A6 with a similar efficacy, their relative recoveries by coimmunoprecipitation should closely match the relative recovery of myc-WT in the primary immunoprecipitation for each transfection condition. The apparent decrease in coimmunoprecipitation of HA-MT relative to HA-WT observed in figure 5D and quantified in figure 5E is not significantly different to that observed for the primary immunoprecipitation of myc-WT observed in figure 5B when cotransfected with HA-WT versus HA-MT. This strongly suggests that the p.R507W mutation does not impair the efficacy of formation of the SLC26A6 multimeric complex per se. The demonstration of formation of MT/WT heteromultimers can explain the dominant negative effect of the MT transporter if it is misfolded, has altered stability in the plasma membrane and/or has reduced oxalate transport ability.
Identification of additional deleterious Slc26a6 mutations in cases with nl from UK Biobank
We next analysed up to 200 619 samples with available exome sequencing data in UK Biobank. Unfortunately, NL was not well recorded in the UK Biobank phenotypes as we identified 5267 patients with recorded NL that corresponds to a prevalence of 2% only. This prevalence is very far from the well-known prevalence of NL in the UK (13%).25 Following a case-only design, we found two heterozygous protein-truncating SLC26A6 mutations (NM_022911.3:c. 3G>A/p.Met1? and NM_022911.3:c.1132C>T/p.Gln378*) in two males. These two mutations were very rare in GnomAD (with a minor allele frequency <0.00005). Those two additional independent cases support a possible contribution of SLC26A6 loss of function mutations to NL. Unfortunately, urinary oxalate excretion is not among the urine parameters available in the UK Biobank phenotype.
Discussion
We show complementary genetic and functional evidence for a novel mechanism of inherited enteric hyperoxaluria in humans. Using a candidate gene analysis of whole-exome sequencing data, we identified a rare c.1519C>T mutation in the SLC26A6 gene encoding an oxalate transporter in a family affected by hyperoxaluria. This mutation leads to the substitution of the arginine in position 507 by a tryptophan. Further extensive in vitro characterisation revealed that the mutation decreases both SLC26A6 transport function and membrane expression. Moreover, the mutant allele also exerts a profound dominant negative effect on the WT allele, suggesting that the mutation might affect SLC26A6-mediated oxalate secretion more than gene haploinsufficiency alone would do.
The results of our experiments demonstrating that coexpression of mutant and WT SLC26A6 decreases total and cell-surface expression of the WT protein, that mutant SLC26A6 is capable of forming multimeric structures with WT SLC26A6 and that the presence of a tryptophan in position 507 is predicted to destabilise the interaction of the mutant SLC26A6 monomer with the lipid bilayer suggest that potential mutant–mutant homodimers and mutant–wild type heterodimers are both very likely to be unstable in the cell plasma membrane. The dominant-negative effect of the mutant SLC26A6 on the wild-type protein assembled in the heterodimer (WT/MT SLC26A6) combined with the inhibitory effect of the mutation itself expressed in the homodimer (MT/MT SLC26A6) would be expected to decrease the production of functional SLC26A6 in the plasma membrane by much more than the 50% predicted by the heterozygous mutation.
SLC26A6 is an anion exchanger from the solute carrier family 26 expressed at the apical membrane of many types of epithelial cells including enterocytes in the gastrointestinal tract.26 It can exchange intracellular oxalate for external chloride, and hence, performs apical oxalate secretion.27 Oxalate transport across the intestinal epithelium results from the combination of passive paracellular absorption and active transcellular secretion.28 Mouse studies have shown that SLC26A6 plays a critical role in intestinal secretion of oxalate.12 13 Therefore, inactivation of Slc26a6 in mice enhances net oxalate absorption, leading to higher plasma oxalate and ultimately increased urinary oxalate excretion, particularly when dietary intake of oxalate is high.12 Our findings suggest that the c.1519C>T/p.R507W mutation in SLC26A6 may cause similar pathophysiology resulting in enteric hyperoxaluria in humans. In this regard, it is striking that the patient’s hyperoxaluria was dramatically reduced by adding calcium to her diet, strongly suggesting that hyperoxaluria in her case was mostly from enteric origin and can be controlled by treatments that limit passive paracellular absorption of oxalate.
Four previous studies have assessed the possible association of SLC26A6 mutations with hyperoxaluria and calcium oxalate urolithiasis:
In the first study, the authors screened a cohort of 94 patients with PH and 96 controls.14 The non-synonymous SLC26A6 variants that were detected in cases were actually frequent (minor allele frequency ≥1%) in one or more populations from the GnomAD browser (v2.1.1). A c.616G>A (p.V206M) mutant was most common (11%) and showed a 30% reduction in oxalate transport activity. However, heterozygosity for this variant did not affect plasma or urine oxalate levels in the study population. There was no significant effect of any identified variants on oxalate excretion. The p.V206M variant of SLC26A6 was subsequently described in a second study.15 This variant was highly frequent (minor allele frequency (MAF) ~10% in all populations). Again, this variant was not associated with urolithiasis or with hyperoxaluria. In a third study, Lu et al16 functionally assessed non-synonymous SLC26A6 variants that were listed in the public database dbSNP, according to in silico analyses. The authors found a significant association between the low-frequency (but not rare) rs184187143 variant (with a MAF of 0.4% in the Finnish population) and urolithiasis risk (unadjusted p value=0.007). However, no mechanistic insight potentially explaining this association was provided. Finally, two additional heterozygous variants of SLC26A6 have recently been reported.17 The first rare p.R621G variant was identified in a non-stone former individual who exhibited a normal (<40 mg/d) urinary excretion of oxalate. According to several in silico programmes (PolyPhen, SIFT, Mutation Taster, Align GVGD), this mutation was totally benign. The second rare p.D674N variant that abolished SLC26A6 expression and Cl−-dependent bicarbonate transport when transfected in HEK cells was identified in a patient presenting with calcium oxalate urolithiasis. However, the authors did not report whether this variant was cosegregating with lithiasis in the family of the patient. Moreover, in the proband the mutation was not associated with hyperoxaluria but rather with a marked hypocitraturia, in line with previous studies from same group showing that mutation of the STAS domain of SLC26A6 can provoke hypocitraturia, another important risk factor for NL, by altering regulation of the citrate transporter NaDC-1.29 30 A very mild hypocitraturia, along with the marked hyperoxaluria, was also observed in our patient. It is very unlikely that such a mild decrease in urinary citrate excretion could cause such a high-frequency recurrent stone disease course, but it may have contributed along with hyperoxaluria to the increased risk for NL.
In summary, our observation that mutation p.R507W is associated with hyperoxaluria in humans supports the hypothesis that like in the mouse SLC26A6 plays a critical role in intestinal secretion of oxalate, thereby limiting net absorption of dietary oxalate. Consistent with this mechanistic hypothesis, we observed a beneficial effect of increasing calcium in the patient’s diet to reduce enteric oxalate absorption and thereby urinary oxalate excretion. Accordingly, other mutations leading to defects in function of oxalate transporter SLC26A6 could be responsible for inherited form of enteric hyperoxaluria and NL in human patients. Therefore, screening for mutations in SLC26A6 should be performed in patients with hyperoxaluria in whom no mutations in the PH disease genes AGXT, or GRHPR or HOGA are found, or in patients with enteric hyperoxaluria who have no evidence of intestinal disease.
Supplemental material
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Institutional review board of the University Hospital of La Réunion (ID :2019/CHU/10). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We would like to thank the patient and her family for their kind help and participation in the study. We would also like to thank Frédéric Allegaert and Nicolas Larcher for expert technical assistance. This research has been conducted using the UK Biobank Application #67 575.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
NC and RBT are joint first authors.
AB and DE are joint senior authors.
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors NC, RBT, ST, BOV, SK, DWD, SB, LB, ED, EV designed, planed, and conducted experiments. All Authors analyzed and interpreted data. NC, RBT, BOV, RC, PSA and DE designed the study. BOV, RC, PSA, AB and DE wrote the manuscript. DE is the guarantor of the work.
Funding This work is funded by the National Centre for Precision Diabetic Medicine – PreciDIAB. PreciDIAB is jointly supported by the French Government under the "Investissement d’avenir" (Investments for the Future) programme managed by the "Agence Nationale de la Recherche" (ANR, French National Agency for Research) (reference: ANR-18-IBHU-0001), by the European Union through the « Fonds Européen de Développement Régional » (FEDER), by the "Conseil Régional des Hauts-de-France" (Hauts-de-France Regional Council) and by the "Métropole Européenne de Lille" (MEL, European Metropolis of Lille). This work was also supported by grants from the French National Research Agency (ANR-10-LABX-46 ((European Genomics Institute for Diabetes)) and ANR-10-EQPX-07–01 (LIGAN-PM)), from the European Research Council (ERC Reg-Seq – 715575), from FEDER and from the "Région Nord Pas-de-Calais". DE and NC are funded by grants from the foundation Philancia and from l’Association pour l’Utilisation du Rein Artificiel à la Réunion. RC is funded by grant from the French National Research Agency (ANR-16-CE14-0031-01 (PROSTARGET)). Support was also provided by NIH grant R01DK033793 to PSA. In addition, SB and LB received funding from the thematic network grant TRENAL of the Deutscher Akademischer Austauschdienst.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.