Article Text

Abstract
Background Minichromosomal maintenance (MCM) complex components 2, 4, 5 and 6 have been linked to human disease with phenotypes including microcephaly and intellectual disability. The MCM complex has DNA helicase activity and is thereby important for the initiation and elongation of the replication fork and highly expressed in proliferating neural stem cells.
Methods Whole-exome sequencing was applied to identify the genetic cause underlying the neurodevelopmental disease of the index family. The expression pattern of Mcm7 was characterised by performing quantitative real-time PCR, in situ hybridisation and immunostaining. To prove the disease-causative nature of identified MCM7, a proof-of-principle experiment was performed.
Results We reported that the homozygous missense variant c.793G>A/p.A265T (g.7:99695841C>T, NM_005916.4) in MCM7 was associated with autosomal recessive primary microcephaly (MCPH), severe intellectual disability and behavioural abnormalities in a consanguineous pedigree with three affected individuals. We found concordance between the spatiotemporal expression pattern of Mcm7 in mice and a proliferative state: Mcm7 expression was higher in early mouse developmental stages and in proliferative zones of the brain. Accordingly, Mcm7/MCM7 levels were detectable particularly in undifferentiated mouse embryonal stem cells and human induced pluripotent stem cells compared with differentiated neurons. We further demonstrate that the downregulation of Mcm7 in mouse neuroblastoma cells reduces cell viability and proliferation, and, as a proof-of-concept, that this is counterbalanced by the overexpression of wild-type but not mutant MCM7.
Conclusion We report mutations of MCM7 as a novel cause of autosomal recessive MCPH and intellectual disability and highlight the crucial function of MCM7 in nervous system development.
- mutation
- missense
- DNA replication
- genetic association studies
- nervous system diseases
Data availability statement
Data are available upon reasonable request. Data may be obtained from a third party and are not publicly available. All data relevant to the study are included in the article or uploaded as supplemental information. Patient data relevant to the study are included in the article. Further experimental data are available from Ethiraj Ravindran (ethiraj.ravindran@charite.de) and genetic data are available from Hao Hu (huh@cougarlab.org) upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The minichromosomal maintenance (MCM) complex with its six subunits MCM2–7 is key for cell proliferation.1 Together with the origin recognition complex (ORC) subunits 1–6 and the licensing factors Cdc6 and Cdt1, it assembles in a prereplicative complex (pre-RC) to initiate DNA replication.2 3 Here, MCM2–7 proteins play an important role in the initiation and elongation of the replication forks by unwinding double-stranded DNA during S phase.4 After the S phase, the pre-RC activity is negatively regulated by phosphorylation of the licensing factors, which in turn prevents the complex to reassemble.5 When this replication process is impaired, neural stem cells (NSCs) are unable to proliferate rapidly, resulting in a decreased progenitor pool, a reduction in the final number of neurons in the brain and ultimately in microcephaly.6–8
Mutations in various components of the MCM complex and other factors involved in DNA replication have been linked to human disease (online supplemental table S1). Heterozygous MCM2 missense variants were identified in one pedigree with eight individuals affected by autosomal dominant deafness 70 (MIM#616968).9 Similarly, the deletion of Mcm2 produced a microcephaly phenotype in zebrafish, and MCM2 downregulation resulted in short cilia and centriole overduplication in human fibroblasts.10 Biallelic MCM4 variants cause immunodeficiency 54 (MIM#609981) with decreased natural killer cells, adrenal insufficiency, prenatal and postnatal growth retardations and also microcephaly.11 Moreover, biallelic MCM5 variants have been reported to cause Meier-Gorlin syndrome 8 (MIM#617564), a disease characterised by microcephaly, prenatal and postnatal growth retardations, microtia and aplasia/hypoplasia of the patellae.12 MCM5 dysfunction caused S-phase progression delay in patient lymphoblastoid and primary skin fibroblasts, which in turn caused impaired DNA replication due to hypersensitivity to replicative stress.12 Variants in MCM7 have been linked to acute myeloid leukaemia due to increased proliferation.13 Similarly, mutations in other components of the pre-RC have been linked to human disease, for example, biallelic ORC1 mutations cause Meier-Gorlin syndrome 1 (MIM#224690) and corresponding patient cells showed impaired replication licensing with slower cell cycle progression and growth restriction.14 Thus, proper DNA replication is required for the accurate proliferation and differentiation, which are necessary to avoid cortical malformations like microcephaly.15
Supplemental material
In this study, we report biallelic mutations of MCM7 as a novel cause of a neurodevelopmental disorder with autosomal recessive primary microcephaly (MCPH) and intellectual disability and further elucidate the role of MCM7 in brain development.
Subjects and methods
Genetic analyses
Detailed genetic methods including whole-exome sequencing (WES) and bioinformatics analysis are described in the online supplemental data.
Prediction of protein structures and mutant (Mut) stability
Domains of MCM7 were identified based on the National Center for Biotechnology Information (NCBI) Conserved Domain Database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi).16 Multiple sequence alignment of MCM7 was performed using the ClustalW program.17 Three-dimensional structural models of MCM7 (MCM7-WT and MCM7-A265T) were predicted by the Swiss-model web tool (http://swissmodel.expasy.org/interactive).18 The visual representation of models and structural superposition were generated by software package Visual Molecular Dynamics.19 The mutant stability change or ΔΔG of variants of MCM7 was predicted using the I-Mutant server (http://gpcr.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi).20
Quantitative real-time PCR (qPCR)
Established methods reported previously were performed for RNA extraction and cDNA synthesis.21 Sets of primers were designed using Primer3 online software (www.primer3.ut.ee) in order to detect Mcm7 cDNA and to obtain specific amplification. Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific, Braunschweig, Germany) according to the manufacturer’s protocol with primers specified in online supplemental table S2 were used for qPCR experiments, and all experiments were run in triplicate. Quantification of the qPCR results was performed as previously described,21 and statistical analysis was performed on GraphPad Prism V.5 Software (GraphPad Software, La Jolla, California, USA).
Cell culture
Mouse embryonal stem cells (mESCs) were cultured and neuronal differentiation was obtained as previously described.22 Human induced pluripotent stem cells (iPSCs) were obtained from the Charité Stem Cell Core Facility. Mouse neuroblastoma (N2a) cells were cultured and maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM GlutaMAX supplement pyruvate; Gibco, Paisley, Scotland) with 10% heat-inactivated fetal bovine serum (FBS, Gibco) and 1% penicillin streptomycin (P/S; Gibco, Grand Island, USA). Cells were maintained at 37°C and passaged at 80% confluence by trypsinisation using 0.05% trypsin/0.02% EDTA for 30 s.
Generation of wild-type MCM7 (WT-MCM7) and mutant MCM7 (Mut-MCM7) overexpression plasmids
We obtained a human WT-MCM7 plasmid from OriGene (OriGene Technologies GmbH, Herford, Germany). The lyophilised plasmid was dissolved and transformed into TOP10 cells (Invitrogen, California, USA). The obtained plasmid was sequenced using the primers listed in online supplemental table S2 to confirm the intact human WT-MCM7 cDNA. We used the WT-MCM7 plasmid as a template to generate a Mut-MCM7 cDNA carrying the same mutation as our index patients (c.793G>A). Site-directed mutagenesis was performed using the QuickChange II XL Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, USA) according to manufacturer’s protocol using MCM7 Mut forward and reverse primers (online supplemental table S2). The PCR reaction was transformed into XL-10 Gold cells, and the obtained plasmids were sequenced using primers listed in online supplemental table S2 to confirm the point mutation.
Generation of WT-MCM7 and Mut-MCM7 overexpressing stable cell lines
MCM7 overexpressing stable cell lines were generated using the WT-MCM7 and Mut-MCM7 overexpression plasmids (OriGene Technologies). Plasmids were linearised using PsiI (New England Biolabs, Frankfurt am Main, Germany), and DNA band elusion was performed using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany) according to the user manual. N2a cells were plated on six-well plates and incubated 48 hours at 37°C. Cells were transfected overnight using Lipofectamine 2000 transfection reagent (Invitrogen) reduced serum medium (Opti-MEM GlutaMAX supplement, Gibco) and 2 µg of linearised overexpression plasmids without antibiotics. Trypsinisation was performed as previously described and cells were plated and incubated overnight at 37°C. Geneticin (600 µg/mL, G-418 Solution; Roche Diagnostics GmbH, Mannheim, Germany) was added to the cells, and the medium was changed every 48 hours for 7 days to obtain only cells with the integrated MCM7 overexpression plasmids.
Quantification of cell viability, apoptosis and proliferation
Cell viability (fluorometric CellTiter-Blue Cell Viability; Promega, Madison, USA), apoptosis (ApoONE Homogeneous Caspase-3/7, Promega) and proliferation assays (colorimetric Cell Proliferation BrdU-ELISA, Roche Diagnostics GmbH) were performed using N2a cells in 96-well plates according to the manufacturer’s protocol, and plates were measured at respective wavelengths using a multiplate reader (SpectraMax iD3; Molecular Devices, San Jose, California, USA). Trypsinisation of N2a cells was performed as previously described, and cells were counted using 0.4% Trypan Blue Solution (Sigma Life Sciences, Steinheim am Albuch, Germany) and seeded at a concentration of 1000 cells/well in 100 µL of DMEM with 5% FBS and 1% P/S per well. Transfection of control siRNA (scramble) (siCo) (AGGUAGUGUAAUCGCCUUG-dTdT) or Mcm7 siRNA (siMcm7) (TGCCAAGCGCTACTCAAGA-dTdT) was performed using Lipofectamine 2000 (Invitrogen) and reduced serum medium, and plates were measured after 48 hours. Statistical analysis for all assays was performed on GraphPad Prism V.5 Software.
Western blot
Protein extraction and Western blot were carried out as previously reported23 with antibodies listed in online supplemental table S3.
Immunohistochemistry
Poly-L-lysine-coated coverslips in 24-well plates or 96-well plates were used to plate mESC/iPSC, which were then fixed in 4% Paraformaldehyde. Cells were incubated in staining buffer (0.2% gelatin, 0.25% Triton X-100, 3% bovine serum albumin) for 30 min in order to permeabilise and block unspecific staining. Primary antibody incubation was done overnight at 4°C, followed by a 2-hour secondary antibody incubation with the antibodies listed in online supplemental table S3. 4’,6-Diamidino-2-phenylindole (DAPI, 1:1000; Sigma-Aldrich) was used for labelling the nuclei. A Zeiss Spinning Disk Confocal Microscope was used to image and analyse the cells labelled with fluorescence, and images were processed using ImageJ.
In situ hybridisation
To obtain a 161 bp Mcm7 PCR product, the primers listed in online supplemental table S2 were used; the PCR product was then purified and cloned into a p-AL2-T vector (Evrogen, Moscow, Russia). The RNA probe was generated by in vitro transcription using T7 or SP6 polymerase (Roche DiagnosticsGmbH), and in situ hybridisation was performed on 16 µm-thick brain sections of mice, stages E14 and P0, as well as whole E10.5 embryos. In situ hybridisation on E14 and P0 brain sections was performed as described previously.24
For whole-mount in situ hybridisation, mouse embryos were fixed in 4% PFA overnight at 4°C, dehydrated and stored in 100% methanol at −20°C. To commence in situ procedure, embryos were rehydrated in decreasing concentrations of methanol (75%, 50% and 25%) for 15 min and subsequently washed in phosphate buffered saline with 0.15% Tween- 20 (PBT). Embryos were then permeabilised at room temperature (RT) using Proteinase K solution (10 µg/mL proteinase K in PBT); the treatment was carried out for 15 min on E10.5 embryos. After digestion, embryos were postfixed in 0.2% glutaraldehyde/4% PFA in PBT for 20 min at RT and washed in PBT. Embryos were incubated in prehybridisation buffer (50% formamide, 5× Saline-Sodium Citrate (SSC) pH 7.0, 2.5 M EDTA, 0.1% Tween-20, 0.15% CHAPS, 0.1 mg/mL heparin, 100 µg/mL yeast tRNA, 50 µg/mL salmon sperm DNA, 1× Denhardt’s solution) at 65°C for 2 hours, after which they were incubated in the desired probe (3 µL probe in 3 mL prehybridisation buffer) at 65°C overnight. Washes of 50% formamide (in 20× SSC), 1 M Tris–HCl pH7.5 in 5M NaCl with RNAse A and Tris buffered saline-Tween-20 (NaCl, KCl, 1 M Tris-HCl pH7.5 and Tween-20) were performed. Embryos were then blocked for 1 hour in 20% sheep serum in TBS-T at RT and incubated overnight at 4°C in anti-Digoxigenin antibody (1:2000) in 5% sheep serum. Washes with TBS-T were performed, followed by alkaline phosphate buffer (5 M NaCl, 1 M Tris–HCl pH 9.5, 1 M MgCl2 and Tween-20) washes. Embryos were placed in chromogenic 5-bromo-4-chloro-3-indolyl-phosphate/nitro bluetetrazolium (NBT/BCIP) (substrate (1:50) in AP buffer until staining developed and reaction was stopped by washes with PBT/1 mM EDTA. Embryos were postfixed with 4% PFA in PBS overnight and stored in 80% glycerol in PBS, at 4°C.
Results
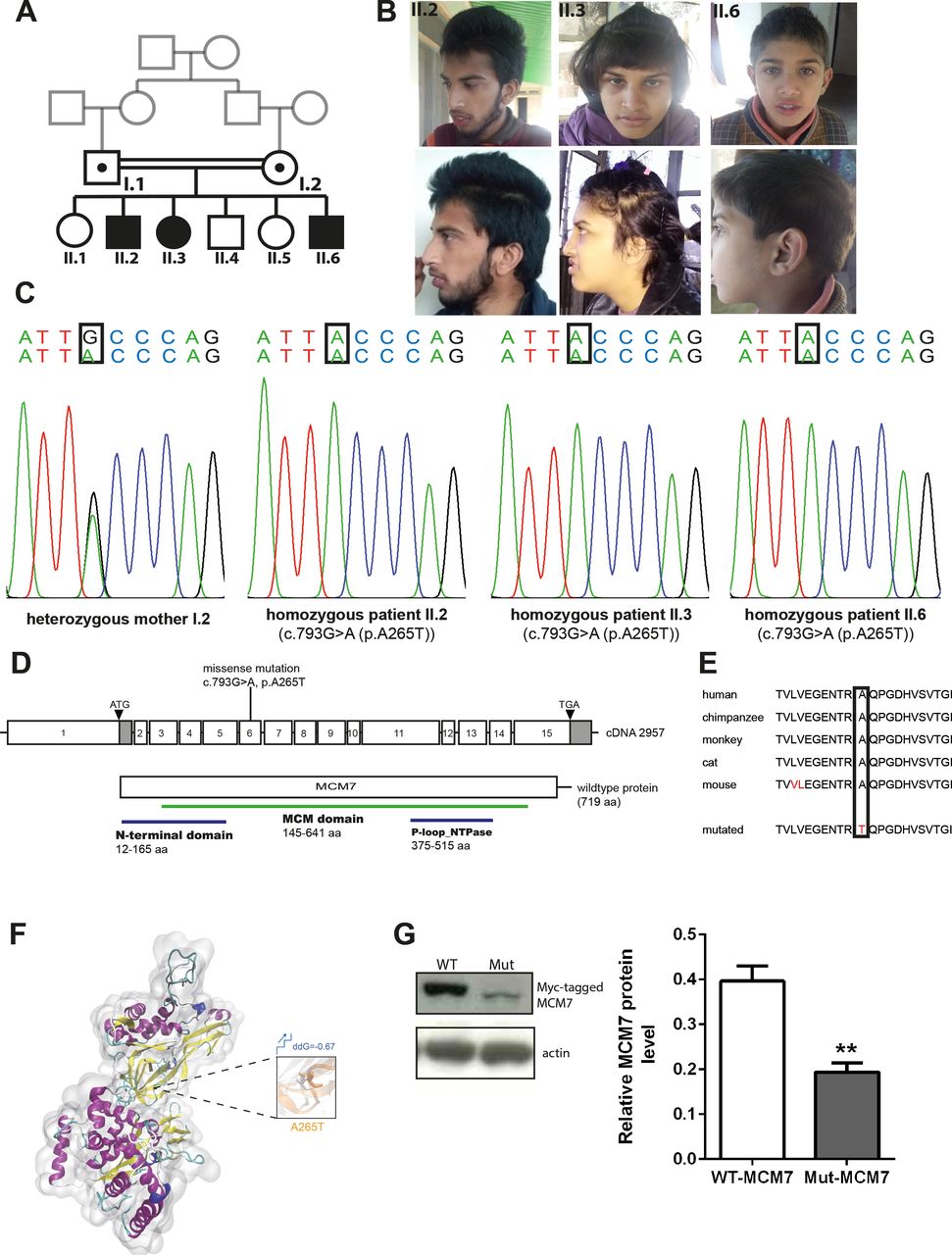
In this study, we report biallelic MCM7 variants as a novel cause of a neurodevelopmental disorder in three offspring of healthy consanguineous parents of Kashmiri–Pakistani descent. All three affected individuals displayed primary (congenital) microcephaly, severe intellectual disability, speech and motor impairments and behavioural abnormalities (table 1) and (figure 1A). Prenatal, perinatal and neonatal medical histories of all the three affected individuals were normal. The affected individuals lacked facial dimorphism (except for II.6, who displayed frontal bossing) (figure 1B) and had no visceral malformations. Microcephaly was severe in all three individuals aged 8 (II.6), 18 (II.3) and 20 (II.2) years at the last examination with head circumference SD ranging from −2.07 in the 8-year-old subject (II.6) to −3.31 in the 20-year-old subject (II.2). Further anthropomorphic data also revealed reduced body weight and length (table 1). The individuals could speak simple words but lacked the ability to formulate complete, even simple sentences. While they were able to walk without support, they were unable to climb up or down the stairs without support. The three subjects were unable to perform routine activities, lacked the concept of self-cleaning, and presented with hyperactivity and aggressive behaviour. They had no clinical or laboratory findings indicative of malignant disease. Results of ophthalmological and otorhinolaryngological examinations were normal.
Clinical features of individuals with biallelic variants in MCM7

Phenotype and genotype of patients with homozygous MCM7 mutation. (A) Pedigree. (B) Pictures of affected individuals. (C) Electropherogram obtained from Sanger sequencing depicting homozygous point mutation in MCM7 c.793G>A (NM_005916.4) in patients II.2, II.3 and II.6, which is heterozygous in the healthy mother. (D) Pictogram of exons 1–15 of MCM7 cDNA with localisation of the patient mutation on exon 6 and structure of the protein with protein domains (N-terminal domain 12–165 aa, MCM domain 145–641 aa, P-loop_NTPase 376–515 aa). (E) The mutation is located in a highly conserved region of the protein (PhyloP 5.059, PhastCons 1). (F) Three-dimensional structural model of MCM7 and close-up view of structural superposition of MCM7-WT (white) and MCM7-A265T (orange), which are displayed with transparent new cartoon representation. The Ala265 and Thr265 residues are shown with white and orange licorice representation, respectively. The change of Gibbs free-energy gap and the stability on mutation are also indicated. (G) Myc-tagged MCM7 protein levels were decreased on a Western blot of N2a cells carrying overexpression of the patients’ mutation (Mut-MCM7) compared with WT-MCM7 overexpressed N2a cells (MCM7 (79 kDa), actin (43 kDa); n=3, unpaired t-test, **(p=0.0066)). MCM, minichromosomal maintenance; Mut-MCM7, mutant MCM7; WT-MCM7, wild-type MCM7.
To identify the genetic cause of the disease phenotype, we performed WES in the index family. Sequencing data revealed the homozygous missense variant c.793G>A (g.7:99695841C>T, NM_005916.4) in MCM7 in all three patients, which was further confirmed by Sanger sequencing (figure 1C,D). The variant segregates with the phenotype in the index family, and no disease-causative variants in other genes previously linked to neurological diseases were identified. The identified variant in MCM7 is located in a highly conserved area and predicted to cause an exchange of a highly conserved hydrophilic nonpolar alanine by a hydrophilic polar threonine at the protein level (p.A265T, NP_005907.3) (figure 1E). Structural analysis of Mut-MCM7 protein revealed that the identified mutation (p.A265T) destroyed the intramolecular hydrophobic interactions with adjacent hydrophobic residues Val271 and Val304 of MCM7 protein, which might impair the local secondary structure and molecular functions (figure 1F). The mutation is disease-causative in nature, as predicted by Mutation Taster, PolyPhen-2 and SIFT (online supplemental table S4) (www.mutationtaster.com, http://genetics.bwh.harvard.edu/pph2, https://sift.bii.a-star.edu.sg).25–27 Protein immunoblot of N2a cells overexpressing Myc-tagged MCM7 carrying the patient mutation p.A265T revealed strongly reduced protein levels when compared with N2a cells in which Myc-tagged WT-MCM7 was overexpressed (n=3, unpaired t-test, p=0.0066) (figure 1G).
Because the MCM complex is known to be a major component in the replication process and proliferation is one of the major processes involved in the early stages of brain development,28 29 we analysed the temporospatial expression of Mcm7 in mice by performing qPCR of Mcm7 mRNA at embryonic (E9, E10, E11 and E14) and postnatal (P0 and P5) stages, and whole-mount in situ hybridisation of E10.5 mouse embryos (figure 2A,B). Mcm7 expression was significantly higher during developmental stages corresponding to proliferation and neurogenesis (E9–E14), as opposed to later postnatal developmental stages (P0 and P5) (figure 2B). In situ hybridisation of E10.5 embryos revealed a ubiquitous Mcm7 expression in the mouse embryo (figure 2A). Based on the interim results, in situ hybridisation was also carried out on E14 and P0 mouse brain sections to understand the spatiotemporal expression pattern of Mcm7. The results revealed a high expression of Mcm7 in the ventricular and subventricular zones of E14 brain sections, consistent with the highly proliferative zones, and reduced expression was present in the neocortex of P0 brain sections, where mainly postmitotic cells reside (figure 2C). Immunohistochemistry was then carried out to analyse the localisation of Mcm7 across cell cycle stages on E14 mouse brain sections. Mcm7 protein colocalised with DAPI, a marker of DNA nuclei, throughout the cell cycle but was dispersed throughout the cytosol during mitotic phases when the chromosomes were condensed (figure 2D).
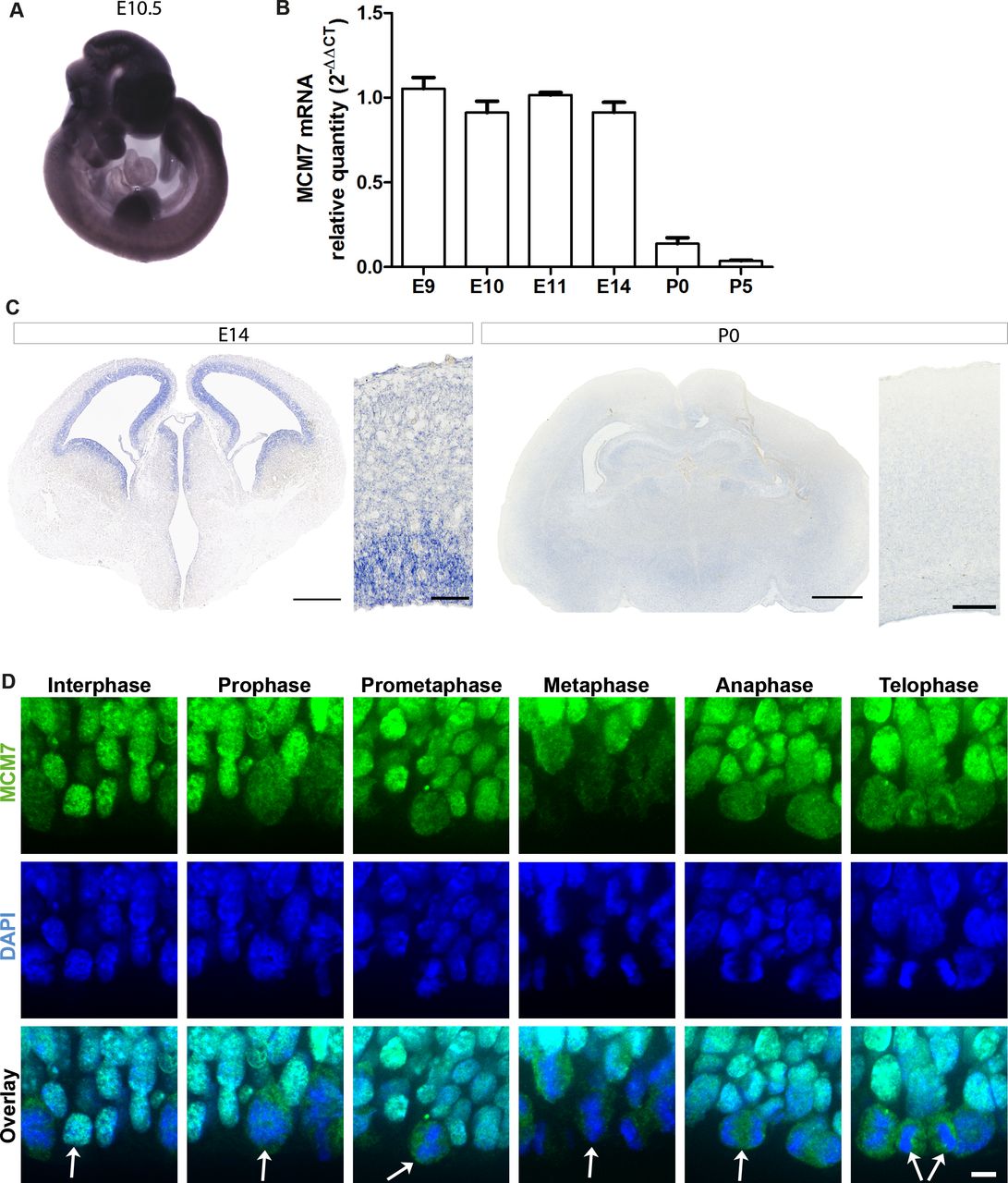
Temporospatial expression of Mcm7 in mouse developmental stages. (A) Whole-mount in situ hybridisation of Mcm7 on E10.5 wild-type mouse embryos revealed ubiquitous expression. (B) Mcm7 mRNA levels analysed by quantitative real-time PCR in mouse embryonal stages E9–E14 and postnatal stages P0 and P5 (n=6 per group). Mcm7 was highly expressed during embryonal stages corresponding to proliferation and neurogenesis with decreased levels seen in postnatal stages. (C) E14 mouse brain sections (scale bar 500 µm/50 µm) showed high expression levels of Mcm7 in the ventricular and subventricular zones, and reduced levels at P0 through in situ hybridisation (scale bar 650 µm/250 µm). (D) Immunostaining on E14 mouse brain sections for Mcm7 (green) and DAPI (blue) revealed the presence of Mcm7 throughout mitosis (scale bar 10 µm). Mcm7 colocalised with DAPI-stained nuclei is dispersed throughout the cytosol in mitosis, when the chromosomes are condensed. Arrows indicate the dividing cells across different stages of mitosis. DAPI, 4’,6-diamidino-2-phenylindole.
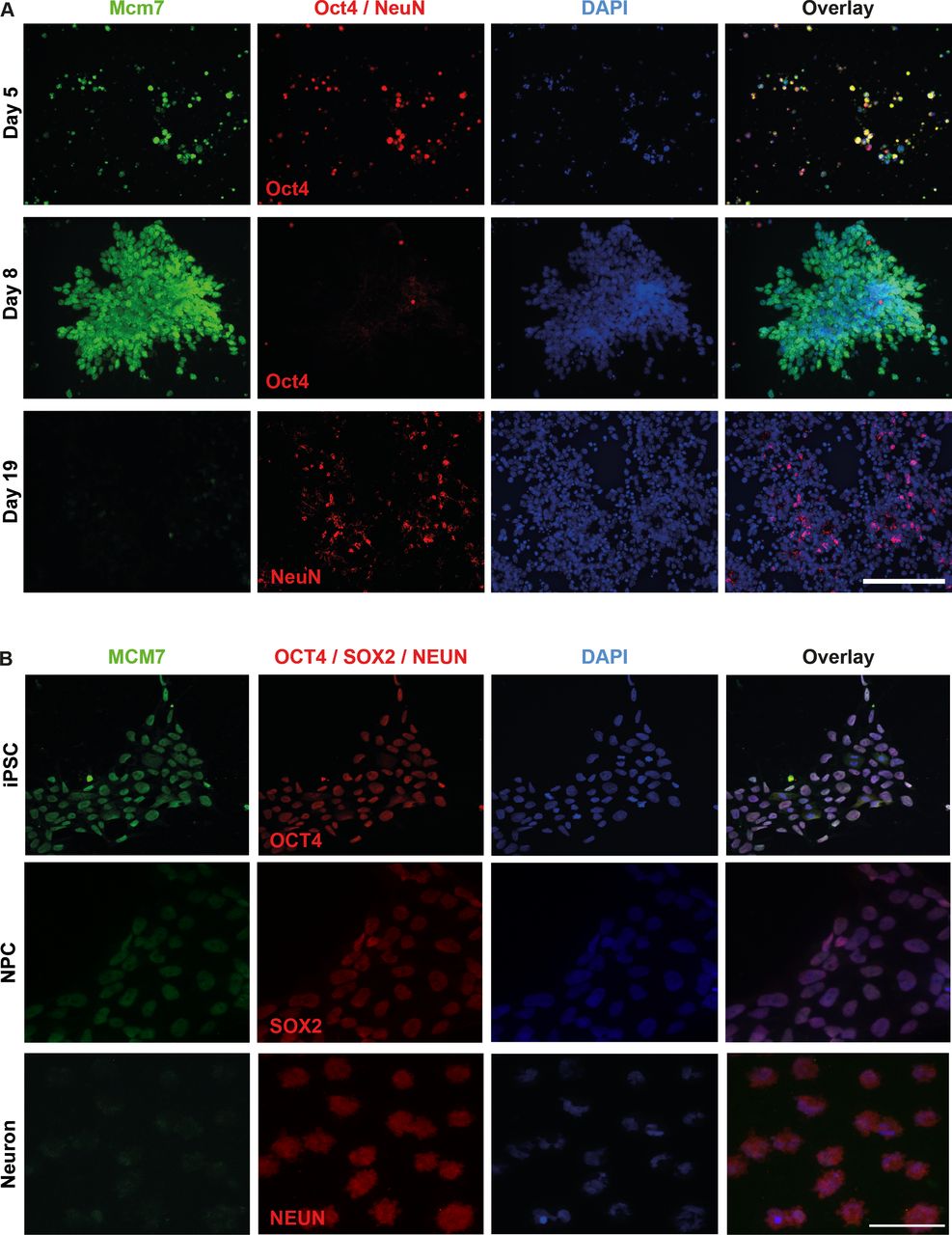
To obtain a better insight into the expression and localisation of Mcm7/MCM7 at different stages of mouse and human cells, we performed immunolocalisation experiments in mESCs as well as human iPSCs and differentiated neurons. Undifferentiated mESCs were costained with Mcm7 antibody on days 5 and 8 using Oct4 as a stem cell marker, and differentiated cells were costained on day 19 using NeuN as a marker for neurons. Increased intensity of Mcm7 staining was observed from day 5 to 8, followed by a significant reduction at day 19, where almost no Mcm7-positive cells were observed (figure 3A). In the case of human iPSCs, staining was carried out for MCM7 on iPSCs, neural precursor cells (NPCs) and differentiated neurons using OCT4, SOX2 and NeuN as markers for these different cell types, respectively. In line with mESC data, reduced intensity of MCM7 was observed in neurons as compared with iPSCs and NPCs, indicating the role of MCM7 in proliferating cells (figure 3B).

Increased Mcm7/MCM7 staining intensity in stem cells compared with differentiated neurons. (A) Costaining of Mcm7 (green) and Oct4 (red) in day 5 mouse embryonic stem cells and day 8 mouse neural stem cells revealed high expression of Mcm7 in undifferentiated cells, whereas differentiated neurons costained with Mcm7 (green) and NeuN (red) on day 19 showed significantly reduced levels of Mcm7 (scale bar 100 µm). (B) Human iPSCs and iPSC-derived NPC costained with MCM7 and OCT4/SOX2 revealed high levels of MCM7, whereas neurons derived from iPSCs and costained with MCM7 (green) and NeuN (red) show greatly reduced intensity of MCM7 (scale bar 50 µm). DAPI, 4’,6-diamidino-2-phenylindole; iPSC, induced pluripotent stem cell; NPC, neural precursor cell.
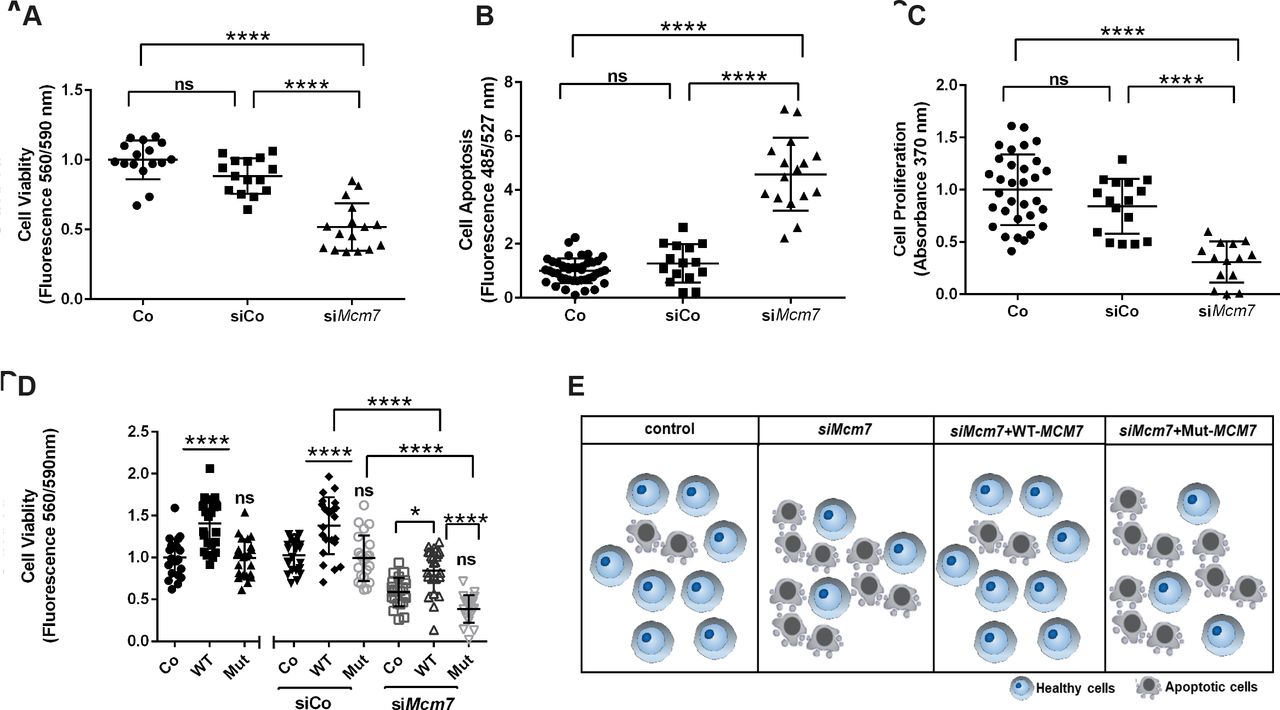
To study whether Mcm7 downregulation has an effect on cell viability and proliferation, we performed corresponding assays on N2a cells treated with either siMcm7 to knockdown Mcm7, scramble siRNA (siCo) or lipofectamine control (Co). Cell viability was significantly decreased in the cells treated with siMcm7 compared with Co and siCo (n=16 per condition, one-way analysis of variance (ANOVA), p<0.0001, Tukey’s multiple comparison test) (figure 4A). Also, siMcm7-transfected cells underwent significantly increased apoptosis and also showed reduced proliferation in comparison to Co and siCo (n=16 per condition, one-way ANOVA; p<0.0001, p<0.001 and p<0.05; Tukey’s multiple comparison test) (figure 4B,C). In all three experiments, no significant difference was observed between Co and siCo (n=16 per condition, one-way ANOVA, p>0.05, Tukey’s multiple comparisons test). Our downregulation experiments confirmed that the knockdown of Mcm7 leads to reduced viability and proliferation with increased apoptosis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Downregulation of Mcm7 reduces cell viability and proliferation and rescued by WT-MCM7 but not Mut MCM7. (A) Cell viability in live N2a cells was significantly impaired when transfected with siMcm7 (one-way ANOVA, ****(p<0.0001), Tukey’s multiple comparison test). (B) N2a cells transfected with siMcm7 showed a higher rate of apoptosis (one-way ANOVA, ****(p<0.0001), Tukey’s multiple comparison test). (C) Proliferation assay revealed a significant decrease of cell proliferation in N2a cells transfected with siMcm7 (one-way ANOVA, *(p<0.05), Tukey’s multiple comparison test). (D) The reduced cell viability phenotype caused by siMcm7 downregulation could be rescued significantly by the overexpressed WT-MCM7 cell line, whereas the Mut-MCM7 could not rescue the phenotype as observed in the control condition (n=24 per condition, one-way ANOVA; ****(p<0.0001), *(p<0.05), Tukey’s multiple comparison test). (E) Pictogram depicting the proof-of-principle: reduced cell viability on knockdown of Mcm7 could be rescued by overexpressed WT-MCM7 but not Mut. ANOVA, analysis of variance; Mut, mutant; ns, not significant; WT-MCM7, wild-type MCM7.
As a proof-of-principle experiment, that is, to confirm that the MCM7 variant identified in the index patients is disease-causative, we performed rescue experiments with WT-MCM7 and Mut-MCM7 overexpressed cell lines. We generated Myc-tagged WT and Mut-MCM7 overexpression plasmids, where the Mut-MCM7 plasmid carried the same variant identified in our patients (c.793G>A). These overexpression plasmids were then used to generate stable WT-MCM7 and Mut-MCM7 overexpressing N2a cell lines, where the overexpression was confirmed by Western blot (figure 1G). Cell viability assays were subsequently repeated using the same transfection conditions mentioned previously (Co, siCo and siMcm7) and each of the cell lines (Co, WT-MCM7 and Mut-MCM7) (figure 4D). The reduced cell viability phenotype caused by siMcm7 downregulation could be rescued significantly in the overexpressed WT-MCM7 cell line, whereas the Mut-MCM7 could not rescue the phenotype observed in the control condition (n=24 per condition, one-way ANOVA, p<0.0001 and p<0.05, Tukey’s multiple comparison test) (figure 4E).
Discussion
Here, we report that biallelic variants in MCM7, a gene known to play a role in DNA replication and therefore important for cell proliferation, result in MCPH with intellectual disability. The MCM7 phenotype classifies as a novel entity of autosomal recessive MCPH, a rare disorder characterised by severe microcephaly at birth and intellectual disability in the absence of visceral malformations. To date, 25 MCPH subtypes caused by variants in 25 loci have been reported (MCPH1–25).30 31 MCPH genes are expressed in the neuroepithelium of the ventricular zone, considered the primary germinal zone of the cerebral cortex during cortical neurogenesis, and are therefore involved in the proliferation of neural progenitor cells.32 MCPH genes have been linked to cell cycle checkpoint control, chromosome condensation, microtubule dynamics, DNA replication, DNA damage–response signalling, and centrosome/mitotic spindle function during embryonic neurogenesis.31 A defect in the proliferation of NSCs which causes a premature conversion of symmetric (stem cell pool regeneration) to asymmetric (differentiation promoting) stem cell division is a main etiological factor of MCPH.33 Thus, MCM7 fits well into the pathomechanism spectrum of MCPH.
Our results indicate that the identified MCM7 variant produces a dysfunctional protein, culminating in impaired replication/proliferation and subsequent disturbed brain development and function. This is in line with previous studies which have reported that inaccuracies during DNA replication can lead to genomic instability with a subsequent decrease in proliferating cells associated with brain malformations.15 34 35 In particular, the MCM complex proteins have been reported to play a crucial role in proliferation and neurogenesis during development.15 Thus, it is evident that MCM7 is strongly expressed in the precursor cells compared with differentiated neurons to regulate the DNA replication and proliferation processes. Our expression data of Mcm7 in the early developmental as well as postnatal stage of mouse brain emphasies its importance for cell proliferation. Furthermore, CDT1 and CDC6, which are part of the pre-RC have been shown to be present in higher levels in Embryonic Stem Cells than in differentiated neurons as a means of securing adequate licensing and initiation of DNA replication.36 37 Our findings also align with a previous report on the localisation of Mcm7 in the proliferative zone of Nicotiana benthamiana root tips,38 emphasising the conserved nature of MCM7 protein across species.
In view of the data on other MCPH/microcephaly genes and our findings for MCM7, the MCM7 variant likely causes microcephaly through defective stem cell proliferation leading to a reduced NPC pool which is reflected in decreased number of matured neurons, culminating in microcephaly. One of the other MCPH genes, CDK5RAP2, has also been shown to encode a centrosomal protein highly expressed in the proliferating cells during development. Mutant Cdk5rap2 mice displayed microcephaly due to reduced cell numbers and reduced dendritic arborisation, indicating the role of Cdk5rap2 in proliferation, neurogenesis and dendritic arborisation.22 23 39 Intriguingly, MCM7 has been observed to strengthen the interaction between two proteins involved in centrosome cohesion, Cep68 and VLH.40 Moreover, the MCM7 target protein Cep68 interacts with Cep215 and PCNT, two proteins acknowledged to be linked to MCPH.41 Mcm7 was also shown to be involved in cilium formation, with its depletion producing a reduction in the length of primary cilia, which was rescued by human MCM7.10 Homozygous MCM4 mutations have been reported to cause adrenal insufficiency and growth retardation similar to that seen in patients with Microcephalic Primordial Dwarfism (MPD).11 Genes involved in DNA replication similar to MCM7 have been linked to a spectrum of phenotypes ranging from isolated microcephaly to growth retardation syndromes, with disorders including Seckel syndrome, Meier-Gorlin syndrome and MPD disorders.15
Our report expands the genetic spectrum of MCPH and also adds MCM7 to the list of previously identified MCM components associated with neurological disorders. We thereby emphasise its role in brain development and broaden the phenotype spectrum of DNA replication-associated disorders. Further reports on individuals with biallelic MCM7 variants will enable us to understand the phenotype spectrum of MCM7-linked disorders.
Data availability statement
Data are available upon reasonable request. Data may be obtained from a third party and are not publicly available. All data relevant to the study are included in the article or uploaded as supplemental information. Patient data relevant to the study are included in the article. Further experimental data are available from Ethiraj Ravindran (ethiraj.ravindran@charite.de) and genetic data are available from Hao Hu (huh@cougarlab.org) upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
The human study was approved by the local ethics committee of the Charité (approval no. EA1/212/08) and University of Azad Jammu and Kashmir, Muzaffarabad, Pakistan and written informed consent was obtained from the parents of the patients for the molecular genetic analysis, the publication of clinical data, and photos. All animal work was carried out in accordance to the national ethic principles and approved by the local committee (T0344/12).
Acknowledgments
We thank our patients and their families for the contribution. We thank Jessica Fassbender, Lena-Luise Becker, Kathrin Blaesius, Bianca Hartmann, Paraskevi Bessa, Sebastian Rademacher, Britta Eickholt, Aleksandra Rusanova, Mateusz Ambrozkiewicz, Victor Tarabykin, Annika Zink, Judit Küchler, Amjad Shehzad for the technical help and discussion. We thank the Berlin Institute of Health stem cell core facility (Harald Stachelscheid, Judit Küchler) for provision of human iPSC and support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AG and AMK are joint senior authors.
Correction notice This article has been corrected since it was published Online First. The list of authors and affiliations has been amended to include Sami Zaqout. The Collaborators and Acknowledgement statements have also been updated accordingly.
Contributors AMK was responsible for project conception. AG, AW, MH, SM contributed clinical samples by recruiting subjects, gathering patient history, clinical information and written informed consents. HH, NL, and XF performed WES and bioinformatics data analysis, NK performed Sanger sequencing and segregation analysis, SZ performed immunostaining, CG and ER performed all other experiments mentioned in this manuscript. CG, ER and AMK drafted the manuscript that was revised and accepted by all coauthors.
Funding The study was funded by the German Research Foundation (DFG, SFB1315, FOR3005), the Berlin Institute of Health (BIH), the Charité, the Major Medical Collaboration and Innovation Program of Guangzhou Science Technology and Innovation Commission (201604020020), the National Natural Science Foundation of China (81671067, 81974163, and 81701451), the Key-Area Research and Development Program of Guangdong Province (2019B020227001) and the Higher Education Commission (HEC) of Pakistan.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.