Article Text

Abstract
Background The genetic architecture of non-acquired focal epilepsies (NAFEs) becomes increasingly unravelled using genome-wide sequencing datasets. However, it remains to be determined how this emerging knowledge can be translated into a diagnostic setting. To bridge this gap, we assessed the diagnostic outcomes of exome sequencing (ES) in NAFE.
Methods 112 deeply phenotyped patients with NAFE were included in the study. Diagnostic ES was performed, followed by a screen to detect variants of uncertain significance (VUSs) in 15 well-established focal epilepsy genes. Explorative gene prioritisation was used to identify possible novel candidate aetiologies with so far limited evidence for NAFE.
Results ES identified pathogenic or likely pathogenic (ie, diagnostic) variants in 13/112 patients (12%) in the genes DEPDC5, NPRL3, GABRG2, SCN1A, PCDH19 and STX1B. Two pathogenic variants were microdeletions involving NPRL3 and PCDH19. Nine of the 13 diagnostic variants (69%) were found in genes of the GATOR1 complex, a potentially druggable target involved in the mammalian target of rapamycin (mTOR) signalling pathway. In addition, 17 VUSs in focal epilepsy genes and 6 rare variants in candidate genes (MTOR, KCNA2, RBFOX1 and SCN3A) were detected. Five patients with reported variants had double hits in different genes, suggesting a possible (oligogenic) role of multiple rare variants.
Conclusion This study underscores the molecular heterogeneity of NAFE with GATOR1 complex genes representing the by far most relevant genetic aetiology known to date. Although the diagnostic yield is lower compared with severe early-onset epilepsies, the high rate of VUSs and candidate variants suggests a further increase in future years.
- epilepsy and seizures
- genetics
- diagnostics
- neurology
Statistics from Altmetric.com
Introduction
Focal epilepsies, in which seizures originate from a circumscribed brain region within one hemisphere, account for 60% of all epilepsy cases.1 While some of these can be attributed to acquired brain lesions, in one-third, no structural epileptogenic abnormality can be identified.2 These epilepsies are then referred to as non-lesional or non-acquired focal epilepsies (NAFEs). Although the exact aetiology often remains unknown, there is emerging evidence for a considerable genetic contribution to NAFE.3
This is underlined by gene discoveries in families with monogenic forms of this entity, as exemplified by the first identified epilepsy gene, CHRNA4, encoding the α4 subunit of the nicotinic acetylcholine receptor,4 or more recently by the discovery of DEPDC5, NPRL2 and NPRL3, which together form the GATOR1 complex, a negative regulator of the mammalian target of rapamycin (mTOR) pathway.5 6
Owing to the increased availability of genetic data and the rapid advancements in high-throughput sequencing technology, our knowledge about the molecular background of epilepsies is constantly expanding. Large-scale next-generation sequencing (NGS) efforts revealed an enrichment of rare coding variants across all types of non-lesional epilepsies (including NAFE) and thus contributed to elucidate their complex genetic architecture.7–9
However, the interpretation of genetic variants in these gene discovery studies is significantly different from the diagnostic real-life setting, in which rigorous criteria have to be applied to establish a genetic diagnosis. The utility of NGS as a diagnostic tool has predominantly been demonstrated for paediatric and mixed cohorts with ill-defined phenotypes.10–13 In contrast, there is a lack of data evaluating diagnostic ES in clinically well-characterised patients with focal epilepsies. One small-scale diagnostic ES study used a targeted analysis approach in 40 patients with NAFE and a suspected genetic background, resulting in a diagnostic yield of 12.5%.14 Two further studies used narrower panel approaches limited to known focal epilepsy genes, both resulting in hit rates of only less than 2%.15 16 These contrasting findings demonstrate the uncertainty about genetic testing outcomes for this relevant epilepsy subgroup, and there is a strong need to evaluate comprehensive genetic–diagnostic approaches in clinically well-characterised cohorts.
In this study, we intended to contribute data to this knowledge gap and performed diagnostic exome sequencing (ES) in 112 deeply phenotyped patients with NAFE adhering to strict clinical–genetic guidelines. Furthermore, we report the clinical characteristics of patients with genetic diagnoses. Being aware that our current ability to assign definite molecular diagnoses is insufficient, we explored plausible candidate genetic aetiologies for NAFE that need to be replicated in future studies.
Methods
Patient selection
All individuals were seen at the Department of Neurology of the Medical University of Vienna, Austria, prior to enrolment. Included subjects underwent a clinical workup comprising brain MRI and video-electroencephalogram (EEG) monitoring. Inclusion criteria were a clinical diagnosis of focal epilepsy and at least one of the following: (1) positive family history (defined as at least one first-degree or two second-degree relatives with a history of seizures), or (2) seizure onset before the age of 25 years or (3) resistance to antiepileptic drug (AED) treatment as defined by the International League Against Epilepsy (ILAE).17 Exclusion criteria were (1) structural abnormalities on brain MRI (except for hippocampal sclerosis/atrophy) and (2) moderate-to-severe intellectual disability. At recruitment, all patients were at least 18 years old and gave written informed consent.
ES and data analysis
Singleton (proband-only) ES was performed at the Institute of Human Genetics (Helmholtz Centre/Technical University Munich, Germany). Exomes were enriched in solution with SureSelect Human All Exon Kits 50 Mb V5 (Agilent, Santa Clara, California, USA). DNA fragments were sequenced as 100 bp paired-end runs on an Illumina HiSeq2500 system (Illumina, San Diego, California USA).18 The mean average coverage was 155-fold.
The two following bioinformatic pipelines were used:
SAMtools: Reads were aligned to the UCSC human reference assembly (hg19) using BWA V.0.5.8. Single-nucleotide variants (SNVs) and small insertions and deletions (indel) were detected with SAMtools V.0.1.7.
GATK: Reads were aligned to the UCSC human reference assembly (hg19) using BWA V.0.5.8. Duplicates were removed using Picard V.2.5.0. Indel realignment, base quality realignment (GATK V.3.6.0) and variant calling (HaplotypeCaller) were done as recommended by GATK practices.
Variants were filtered based on the minor allele frequency (MAF), which was estimated using our in-house database (>18 000 exome datasets) and confirmed by the Genome Aggregation Database (gnomAD). Variant prioritisation was based on autosomal recessive (MAF<0.1%) and dominant (MAF<0.01%) filters.
Copy number variant (CNV) analysis
Exome-based CNV analysis was done using ExomeDepth19 and Pindel.20 Pathogenic CNVs were confirmed by SNP Array Affymetrix CytoScan 750K Array (Affymetrix, Santa Clara, USA). Scanning was performed by the Affymetrix GeneChip Scanner 3000 7G and data were analysed using the Affymetrix Chromosome Analysis Suite Software V.3.3, hg19.
Molecular diagnoses according to clinical–genetic guidelines (diagnostic variants)
The initial diagnostic analysis step was targeted to 15 established focal epilepsy genes: CHRNA2, CHRNA4, CHRNB2, DEPDC5, GRIN2A, KCNQ2, KCNQ3, KCNT1, LGI1, NPRL2, NPRL3, PRRT2, RELN, SCN2A and SCN8A.21 In a second step, to also account for unexpected genotype–phenotype correlations and phenocopies, we searched for pathogenic and likely pathogenic variants (herein referred to as diagnostic variants) in a comprehensive set of all 455 genes associated with ‘epilepsy’ in the Online Mendelian Inheritance in Man database (online supplementary table S1). The standard criteria of the American College of Medical Genetics and Genomics (ACMG) were applied for variant interpretation.22 Missense variants in GATOR1 complex genes (DEPDC5, NPRL2 and NPRL3) were classified based on MAF (according to gnomAD) and the Mendelian Clinically Applicable Pathogenicity Score (M-CAP) in silico prediction,23 as recently proposed for this genetic subgroup.24
Supplemental material
Variants of uncertain significance (VUSs) in 15 focal epilepsy genes
Apart from diagnostic variants, we also reported variants with a possible but (according to diagnostic standards) unconfirmed pathogenic effect, hence classified as VUSs. This screen was stringently limited to non-synonymous (missense and in-frame indel) variants in the 15 genes with an established association with genetic focal epilepsies.21 We only selected very rare variants with a MAF<0.01%, and we set a combined annotation-dependent depletion (CADD) score threshold of >20 for missense variants to be reported as VUS in a diagnostic setting.
Rare variants in NAFE candidate genes
To exploratively prioritise potential candidate genes, we searched for rare (MAF<0.01%) non-synonymous (missense, indel, splice-site, frameshift and nonsense) variants in each exome dataset. Variants were manually screened using in silico prediction tools (CADD, sorting intolerant from tolerant (SIFT) and PolyPhen-2 (pph2)),25–27 gnomAD gene constraint information (z score, probability of being loss-of-function intolerant score (pLI)). In the second step, we searched for anecdotal evidence in the literature potentially linking genes with detected variants to focal epilepsy. This prioritisation intended to identify promising candidate variants without being comprehensive.
Statistical analyses
The rate of diagnostic (ie, pathogenic and likely pathogenic) variants in the cohort established the diagnostic yield. We further aimed to explore clinical and demographic characteristics (sex, age at seizure onset, history of febrile seizure (FS) and a positive family history for seizures) associated with a molecular diagnosis. Fisher’s exact test was used for categorical data, and Mann-Whitney U-test for continuous data. A p value of <0.05 was considered statistically significant. Correction for multiple testing (eg, Bonferroni) was not applied in this clinically oriented study with a relatively small sample size. SPSS V.25.0 was used for statistical analyses.
Results
Patient characteristics
Of the included patients, 57% were women (n=64) and 43% were men (n=48). The median age at seizure onset was 15 years (range 1–67 years). Fifty-one per cent (n=57) were diagnosed with temporal lobe epilepsy (TLE), followed by frontal lobe epilepsy (FLE) with 24% (n=27), unclassified focal epilepsy (UFE) with 21% (n=24), parietal lobe epilepsy and occipital lobe epilepsy (OLE) with two patients each. An MRI of the brain was normal in 90% (n=101) and showed hippocampal sclerosis/atrophy in 10% (n=11). One-third (n=37) had a positive family history with at least one first-degree or two second-degree relatives being affected with seizures. Ten patients (9%) had a history of FS preceding afebrile focal seizures. At last documented follow-up, the majority of individuals (77%) was resistant to AED treatment as classified by ILAE.17 The main clinical and demographic characteristics of all 112 included patients are displayed in table 1.
Demographic and clinical features of included patients and associations with diagnostic outcome
Diagnostic outcomes
When strictly using ACMG standard criteria, a diagnostic yield of 9% was achieved with heterozygous diagnostic variants identified in 10/112 individuals. The diagnostic yield was increased to 12% (13/112 individuals) after additionally applying the criteria for the interpretation of GATOR1 complex variants, as recently proposed by Baldassari et al.24
Of these 13 diagnostic variants, nine were detected by the initial diagnostic analysis, which was limited to 15 established focal epilepsy genes. Four additional diagnoses (STX1B, SCN1A, PCDH19 and GABRG2) could be made after applying the extended approach analysing all 455 genes (online supplementary table S1).
The detected variants eventually classified as pathogenic or likely pathogenic included five truncating variants (frameshift or nonsense), five missense variants, two pathogenic CNVs (microdeletions) and one splice-site variant (online supplementary figure S1).
Supplemental material
Diagnostic variants were found in six different epilepsy-associated genes (figure 1). Variants in two genes of the GATOR1 complex (DEPDC5 and NPRL3), a negative regulator of the mTOR signalling pathway, together accounted for 9 of the 13 solved cases (69%). Four truncating and two missense variants were found in DEPDC5, while one truncating, one missense variant and one microdeletion were found in (or involving) NPRL3, in line with haploinsufficiency being the known pathomechanism. No genetic diagnosis was related to NPRL2.

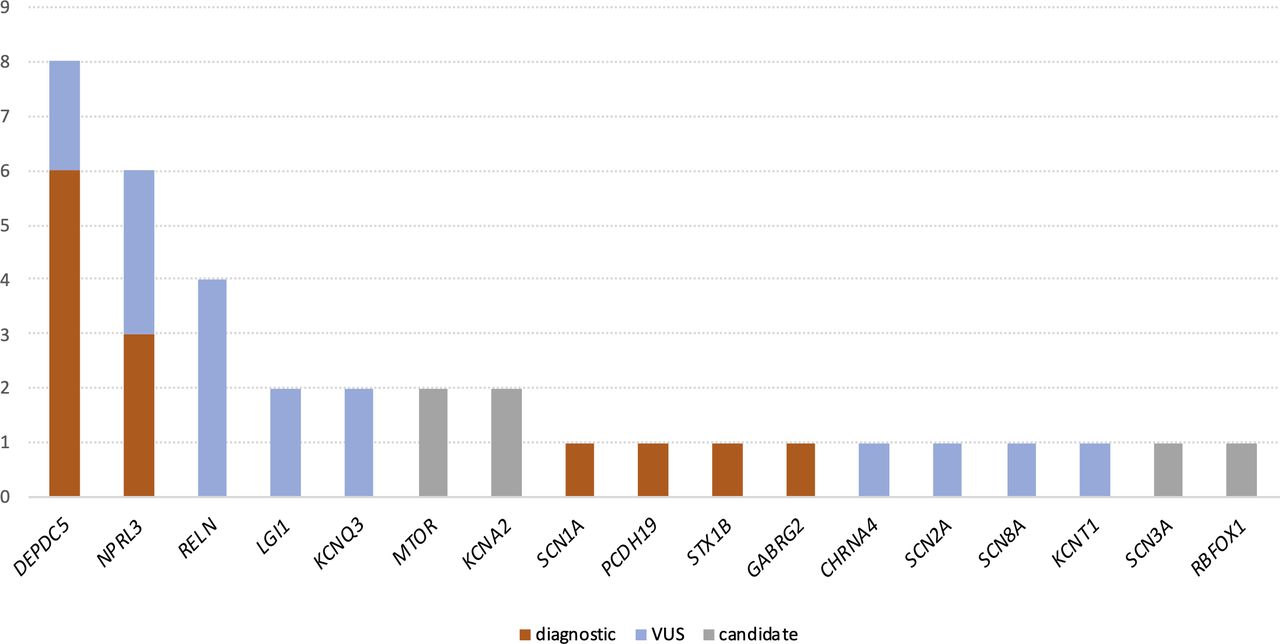{kind=link}
Genes with detected variants in non-acquired focal epilepsy. Number of patients (y-axis) harboring diagnostic variants (orange), VUSs (blue) and candidate genetic aetiologies (grey) reported in this study; genes with detected variants are listed on the x-axis. VUS, variant of uncertain significance.
Patients with an established molecular diagnosis after ES had a younger median age at seizure onset compared with cases without a genetic diagnosis (9 vs 15 years, uncorrected p=0.01). Sex, a positive family history for seizures and a history of FS were not significantly associated with a positive genetic diagnosis (table 1).
Epilepsy phenotypes of patients with a genetic diagnosis
The nine patients with diagnostic variants in the GATOR1 complex genes DEPDC5 and NPRL3 mirrored the broad clinical spectrum of NAFE with a median age of onset of 10 years. In GATOR1-related NAFE, the epilepsy was classified as TLE (n=3), FLE (n=3), UFE (n=2) and OLE (n=1). The family history was positive in six of nine patients. Given the known association with structural brain abnormalities (mainly focal cortical dysplasia), all MRI scans of patients with reported variants in GATOR1 complex genes were re-evaluated by a neuroradiologist, but no focal abnormalities were revealed in retrospect.
We found a likely pathogenic missense variant in GABRG2 (previously reported as a pathogenic de novo variant)28 in a female patient with a history of FS starting during the first month of life. Subsequently, she developed bilateral tonic–clonic seizures and focal seizures with and without impaired awareness. MRI-negative OLE was diagnosed due to the visual seizure semiology and the ictal EEG onset in the occipital region.
One pathogenic CNV was a 3 Mb deletion involving PCDH19 in a female patient diagnosed with MRI-negative FLE, age of 2 years at seizure onset and a negative family history for epilepsy. In contrast to many previously reported patients with this condition, there was no history of FS or cognitive impairment. The clinical course was characterised by severe AED resistance and a high seizure frequency with focal seizures occurring on a daily basis.
The pathogenic missense variant in SCN1A was absent from controls, consistently predicted to be deleterious by multiple in silico tools (pph2, SIFT and CADD) and previously reported to be pathogenic according to ClinVar. The patient harbouring this variant suffered from NAFE with suspected left frontal seizure origin and preceding recurrent (partly complex) FS. Seizures were generally well controlled with valproic acid and levetiracetam, whereas oxcarbazepine and felbamate did not lead to satisfying seizure control.
One male patient with a likely pathogenic splice-site variant in STX1B was affected by severe treatment-refractory childhood-onset TLE. Aside from multiple AED trials, the patient also underwent epilepsy surgery (anteromedial temporal lobe resection). Brain histology revealed a mild malformation of cortical development (mMCD), which was not detectable by MRI. Subsequently, the patient also underwent deep brain stimulation but experienced ongoing seizures when last seen.
Demographic and clinical details of patients with diagnostic variants are delineated in table 2.
Detailed clinical characterisation of all patients with diagnostic variants in known epilepsy genes
VUSs in 15 focal epilepsy genes
Apart from diagnostic variants according to strict clinical standards, we sought to identify further relevant rare variants, that is to say, VUSs in the 15 well-established focal epilepsy genes. We detected 17 VUSs (16 missense variants and 1 in-frame indel) in 9 of the 15 preselected focal epilepsy genes in 16/112 patients. Six of these VUSs were singletons (ie, absent from gnomAD). The reported VUSs are listed in table 3.
Variants of uncertain significance in 15 focal epilepsy genes
Rare variants in NAFE candidate genes
Using an extended exome-wide screen for rare non-synonymous variants, we identified six rare missense variants with damaging in silico prediction in four candidate genes for NAFE with existing but so far only limited evidence for a disease association. This included variants in the genes MTOR (2×), KCNA2 (2×), RBFOX1 (1×) and SCN3A (1×). Details about the prioritised candidate variants are displayed in table 4.
Prioritised candidate variants in genes with anecdotal evidence for an association with non-acquired focal epilepsy
Considering all genetic variants reported in this study (including diagnostic variants, VUSs and candidate variants), 31/112 patients (28%) carried at least one variant.
Multiple rare variants
When also accounting for VUSs and prioritised candidate variants, 5 of 112 patients had double hits in different epilepsy genes. Two of these patients carried one pathogenic variant and one VUS (DEPDC5/RELN and NPRL3/SCN2A), and one patient harboured two VUSs in focal epilepsy genes (LGI1/NPRL3). However, this patient had familial lateral TLE with auditory auras, primarily suggesting a pathogenic role of the LGI1 variant given the characteristic phenotypic constellation. Two further patients each had one VUS and one candidate variant (DEPDC5/SCN3A and RELN/KCNA2).
A comprehensive list including all rare variants with a MAF of <0.01% detected in our cohort is provided in online supplementary table S2.
Supplemental material
Discussion
Over the past few years, large-scale sequencing studies have convincingly demonstrated a burden of rare variants in non-lesional epilepsies, including NAFE.7–9 In spite of this rapid progress, it is still unclear how the emerging knowledge derived from such studies can be translated into the clinical–diagnostic setting. Previous studies evaluating the diagnostic use of NGS were primarily performed in clinically heterogeneous cohorts, often with concomitant syndromic features.10–13 In contrast, there is still a remarkable lack of data addressing the clinical application of NGS for the more common non-lesional epilepsies, of which NAFE is the one most frequently encountered in adult epileptology. To fill this gap, we analysed 112 exomes of well-phenotyped patients with NAFE, aiming to provide a clinical–genetic diagnosis.
Using a broad exome-based approach, we identified molecular diagnoses in 12%, which is less than in syndromic epilepsies,13 but comparable to the small-scale study by Perucca et al evaluating diagnostic ES with targeted gene analysis in NAFE.14 In contrast, our diagnostic yield is remarkably higher than that reported by Hildebrand et al and Tsai et al, who found diagnostic variants in only 0.8% and 1.85% of cases, respectively.15 16 First, the lower hit rate may be explained by the broader inclusion criteria of these studies, not accounting for age of onset and a positive family history. Second, phenotyping was less accurate; for example, some patients did not receive an MRI scan prior to inclusion. Moreover, they did not include CNVs and in-frame indels, which may also play a role in NAFE. The lower diagnostic yield may also partly be explained by the fact that these two studies only included DEPDC5, but not NPRL2 and NPRL3 in their analyses.15 16
Of note, two pathogenic microdeletions were detected in our study, highlighting that monogenic CNVs should also be considered. This is a strong argument in favour of diagnostic approaches combining SNV and CNV analysis. While playing an acknowledged role in the severe infantile epilepsies with comorbid features, we point out that the monogenic role of CNVs in NAFE is probably underestimated.29
In line with previous NGS data, our results demonstrate the implication of a few common genes and a much greater number of rarely mutated genes in the aetiology of NAFE with undistinguishable phenotypes.30 Four of the 13 pathogenic variants were detected in genes with previously no robust evidence for an association with NAFE. However, recent data have suggested that NAFE is also part of the phenotypic spectrum of STX1B and GABRG2 that were previously associated with more severe generalised epilepsies and developmental and epileptic encephalopathies (DEEs).31 32 We hypothesise that in our study, these cases possibly represent phenocopies, since also generalised EEG changes were documented in the patient carrying the GABRG2 variant, and the histological workup eventually revealed an mMCD in the patient with the STX1B variant. This exemplifies the inability to predict the causative genetic defect by the phenotype alone, which is a strong argument in favour of broad genomic testing early in the diagnostic pathway. Given the significant correlation between age at onset and a molecular diagnosis, patients with early-onset NAFE may preferentially be selected. In contrast, somewhat surprisingly, our data (in line with the findings of Perucca et al)14 do not support a positive family history alone as a reliable predictive marker for a genetic diagnosis.
Some of the genetically solved cases in our cohort demonstrate that a molecular diagnosis may have direct management implications for the patients. Despite the genetic heterogeneity, almost 70% of diagnostic hits were detected in genes of the GATOR1 complex (formed by DEPDC5, NPRL2 and NPRL3), pointing towards a major role of the mTOR signalling pathway in the pathogenesis of NAFE. GATOR1 normally suppresses mTOR signalling, so that haploinsufficiency in turn results in mTOR hyperactivation.33 The decisive role of the mTOR pathway is particularly interesting, as pharmacological mTOR inhibition (eg, by everolimus) might specifically be beneficial for this patient population usually characterised by a high rate of AED resistance.34 Furthermore, as opposed to other genetic (eg, SCNA1-related) epilepsies, there is at least anecdotal evidence that these patients may achieve satisfactory seizure outcome following epilepsy surgery.35 These observations together give rise to the hope that effective personalised treatment approaches may become available for this difficult-to-treat patient group. Another example with the potential for a genotype-guided treatment option is the patient with a pathogenic SCN1A variant who did not respond to oxcarbazepine treatment. Having a precise genetic diagnosis at hand after genetic testing, there is now a rational argument to avoid sodium channel blockers.
Apart from diagnostic variants, we found a high rate of VUSs in established focal epilepsy genes and rare deleterious missense variants in four candidate genes (MTOR, KCNA2, RBFOX1 and SCN3A) that have already been associated with NAFE, but for which evidence is limited.36–39 Though not formally classifiable as pathogenic in a diagnostic report, it can be presumed that at least some of these variants are indeed disease relevant (in a monogenic sense). As exemplified by our study, collating well-characterised phenotypes and variants is essential to reveal the clinical significance of genetic defects and to elucidate genotype–phenotype correlations in a stepwise manner. The ongoing expansion of gene discoveries and known variants as well as a periodic reanalysis of exome data have the potential to enhance the diagnostic outcomes in coming years.40
One noteworthy factor possibly limiting the diagnostic yield in our study is the lack of family genotype data, which are useful to confirm segregation in familial cases or de novo mutagenesis in sporadic cases. However, it is widely accepted that the penetrance of variants in epilepsies is often incomplete, meaning that the presence of a variant in a healthy family member does not necessarily exclude disease causation. The exact reason for this phenomenon is still poorly understood but may in part be due to multiple superimposed genetic variants contributing to disease causation in an oligogenic framework. As illustrative examples, we found genetic double hits (including diagnostic variants, VUSs and candidate variants) in five different patients. Acknowledging that this observation is purely explorative and therefore not sufficient to prove an oligogenic or digenic effect, we consider that multiple rare variants may act a part in epileptogenesis. Yet, this hypothesis requires larger sample sizes and functional investigations for confirmation.
Taken together, our data underscore a considerable but heterogeneous genetic contribution to NAFE with GATOR1 complex genes representing the by far most common monogenic aetiology identified to date. Although the diagnostic yield is relatively low compared with early-infantile phenotypes, the high rate of VUSs and emerging candidate aetiologies suggest that outcomes may improve, along with the constantly expanding knowledge. In view of the increased availability of genotype-guided individualised treatments (particularly for GATOR1/mTOR-related epilepsies), ES constitutes a promising diagnostic tool also for this patient population.
Acknowledgments
All authors thank the patients for participating in the study. MK also thanks the Austrian Society of Neurology and the Austrian Society of Epileptology, which supported him with a 6-month research fellowship.
References
Footnotes
Contributors FZ designed and supervised the study. MK analysed exome data and wrote the manuscript. TM supervised the genetic analyses. TMS was responsible for the bioinformatic pipeline. MW and AZ were involved in genetic data analysis. CH was responsible for DNA preparation and quality check. EG, SW and BL-D were involved in the sequencing pipeline. TB was responsible for the secondary confirmation of copy number variants. GK re-evaluated patient MRIs. EP, SA-W and ES were involved in the clinical diagnosis of patients.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study was approved by the ethics committee of the Medical University of Vienna (ethics approval number: 2051/2016).
Provenance and peer review Not commissioned; externally peer reviewed.