Article Text

Abstract
Background Neurofibromatosis type 1 (NF1) predisposes to breast cancer (BC), but no genotype-phenotype correlations have been described.
Methods Constitutional NF1 mutations in 78 patients with NF1 with BC (NF1-BC) were compared with the NF1 Leiden Open Variation Database (n=3432).
Results No cases were observed with whole or partial gene deletions (HR 0.10; 95% CI 0.006 to 1.63; p=0.014, Fisher’s exact test). There were no gross relationships with mutation position. Forty-five (64.3%; HR 6.4–83) of the 70 different mutations were more frequent than expected (p<0.05), while 52 (74.3%; HR 5.3–83) were significant when adjusted for multiple comparisons (adjusted p≤0.125; Benjamini-Hochberg). Higher proportions of both nonsense and missense mutations were also observed (adjusted p=0.254; Benjamini-Hochberg). Ten of the 11 missense cases with known age of BC occurred at <50 years (p=0.041). Eighteen cases had BRCA1/2 testing, revealing one BRCA2 mutation.
Discussion These data strongly support the hypothesis that certain constitutional mutation types, and indeed certain specific variants in NF1 confer different risks of BC. The lack of large deletions and excess of nonsenses and missenses is consistent with gain of function mutations conferring risk of BC, and also that neurofibromin may function as a dimer. The observation that somatic NF1 amplification can occur independently of ERBB2 amplification in sporadic BC supports this concept. A prospective clinical-molecular study of NF1-BC needs to be established to confirm and build on these findings, but regardless of NF1 mutation status patients with NF1-BC warrant testing of other BC-predisposing genes.
- breast cancer
- genotype-phenotype correlation
- neurofibromatosis Type 1
- Nf1 microdeletion
- Brca1/2
Statistics from Altmetric.com
Introduction
Neurofibromatosis type 1 (NF1) is an autosomal dominantly inherited tumour predisposition syndrome caused by constitutional mutations in the NF1 gene, which is diagnosed clinically according to established criteria.1 It has variable expressivity, age-related penetrance and a prevalence of 1/2000–1/3500.2 3 The NF1 gene has 61 exons (4 of which are alternatively spliced) and is responsible for the expression of the 2818 amino acid protein neurofibromin, which acts as a tumour suppressor. Consistent with Knudson’s two-hit hypothesis, in the tumours of patients with NF1 with heterozygous constitutional mutations the wild-type allele is inactivated by somatic mutation. Neurofibromin is a key negative regulator of the Ras signalling pathway and is a Ras-specific GTPase-activating protein (GAP) with sequence and structural homologies to the GAP superprotein family, including a GAP-related domain (GRD).4 Also recognised, but less well characterised, are cysteine-rich/serine-rich (CSRD), tubulin binding, Sec14 homology-like, pleckstrin homology-like and syndecan-2 binding domains (see online supplementary table 1S).
Supplemental material
As the mutations that cause NF1 vary in size from large deletions of >1 Mbp to single bp substitutions in any of the exons, so reported detection rates vary, from 60% to 95% depending on the techniques and tissue source.1 5 Point mutations are observed in all exons and, typical of a tumour suppressor gene, are mostly nulling or protein truncating, while a minority (9.4%–15%) are missense.5 6
NF1 predisposes to many tumour types, both benign and malignant, including breast cancer (BC). BC in NF1 (NF1-BC) usually occurs in women, but has been reported in males, both bilaterally and at a young age.7–9 That there is an increased risk of developing BC in NF1, and in particular that this risk is greater under the age of 50 years, has now been confirmed in a number of studies, with risks of NF1-BC to age 50 years of 7.8%–8.4%, compared with 2% in the general population.8 10 While studies providing age-corrected standardised incidence ratios (SIR) consistently show an increased risk under age 50 years (95% CI 4.0 to 8.8), the most recent also gives an increased SIR over age 50 years (2.0; 95% CI 1.2 to 3.1), consistent with the non-significant estimate published by Wang et al 11 ,8 10–14 (table 1). In addition, two studies have reported increased mortality and unfavourable prognostic factors with NF1-BC.8 15 So, BC tends to occur at a younger age in NF1, and is more malignant.
Summary of studies giving standardised incidence ratios (SIR) of female breast cancer (C50) in NF1
Genotype-phenotype correlations are well described in tumour-predisposing conditions. However, while it has been difficult to establish genotype-phenotype correlations in NF1 some associations have been shown. Patients with large deletions encompassing NF1 and 13 flanking genes (‘NF1 microdeletions’), tend to exhibit a more severe phenotype including learning difficulties and an increased probability of malignant peripheral nerve sheath tumours (MPNST), while an absence of cutaneous neurofibromata is seen with two particular mutations: an in-frame deletion (NF1 c.2970_2972del p.Met992del) and a missense (MS; p.Arg1809Cys).16–20 A high incidence of Noonan syndrome features has been reported in patients with NF1 with missense (MS) mutations at codon 1809, and MS mutations of codons 844–848, that is, within the CSRD, have recently been found to cause a more severe NF1 phenotype including MPNSTs, OPGs and malignant neoplasms in general.18 21 22 Patients with spinal neurofibromas have been found to be more likely to have MS or splice site (SS) mutations and those with optic pathway gliomas are more likely to harbour mutations in the 5′ third of NF1.23 24 While these genotype-phenotype relationships have been found with constitutional mutations, less has been described on somatic NF1 mutations in BC, but a recent review has shown that in sporadic cancers with somatic NF1 mutations BC stands out as the only major cancer type in which amplification of NF1, so gain of function, is as commonly seen as loss-of-function mutations.25 It is also notable that compared with benign neurofibromas large deletions predominate as the somatic mutation observed in MPNSTs and are more likely to extend into 17p (to include the TP53 locus).26 27
Therefore, our aim was to explore potential NF1 genotype-phenotype correlations regarding the risk of BC in NF1, which might aid in clinical care including counselling and, potentially, surveillance or treatment options. Hence, through a worldwide collaboration, we ascertained a set of 78 unrelated NF1-BC cases with defined constitutional NF1 mutations.
Methods
NF1-BC inclusion criteria were: a clinical diagnosis of NF1 (according to NIH criteria), a defined constitutional NF1 mutation and BC at any age. Information was sought on NF1-associated features, the age at BC diagnosis and histological type, other tumours and whether the patient had any other genes tested, but these were not essential.
The constitutional NF1 mutations in the NF1-BC set were determined by standard techniques: direct sequencing of gDNA or cDNA, with testing for microdeletions and intragenic CNV by a combination of MLPA (Multiplex Ligation-dependent Probe Amplification) and array CGH, and defined according to the reference sequence hg19:NM_000267.3.
Reference data on constitutional NF1 mutations were obtained from the NF1 Leiden Open Variation Database (LOVD; accessed 23 April 2018).28 For variants with >1 pathogenicity classification, the more pathogenic was used. Variants not considered pathogenic or probably pathogenic, that is, classes 1–3, were then excluded.
The predicted effect of any given mutation was then defined according to the types in the LOVD, that is, NS (nonsense), FS (frameshift), SS (splicing affected), MS (missense), ID (small deletion/duplications of up to 50 codons; in-frame deletions/duplications; indels), LD (large deletions) or OT (others, including start codon mutations). Care was taken in interpretation of the effect of mutations on the protein as, in particular, some putative MS mutations may also cause splicing abnormalities.5 29 In instances where there was >1 predicted effect, then MS/SS or NS/SS or SS/any were called as SS, and NS/FS as FS. Finally, 10 of the NF1-BC cases were found to have been reported to the LOVD and were therefore excluded from the LOVD set, making n=3432.
Incidence rates of BC in BRCA1/2 and TP53 mutation carriers, and the UK population (ICD-10: C50; 2009–2011) were obtained from published sources.30–32 The statistical significance of associations was determined using Fisher’s exact test, two-tailed given no prior alternative (http://www.langsrud.com/stat/fisher.htm), with adjustment for multiple comparisons using the p value plot method of Schweder and Spjøtvoll, and the Benjamini-Hochberg procedure (www.biostathandbook.com/multiplecomparisons.html).33 34
Results
Clinical cases
The NF1-BC cohort (ascertained 2013–2017) consists of 78 unrelated NF1 cases with BC of which 75 were female (table 2). Multiple BCs occurred in six cases (8.3%): bilaterally in one male and three females (two of them metachronously), plus two other cases of synchronous BC in females. Information on type was available for 31 (40%): 29 (94%) were ductal, 2 (6%) were lobular.
Clinical and molecular details of patients with NF1 and breast cancer
Eighteen NF1-BC cases had undergone testing of BRCA1/2, of which seven had also had TP53 analysis. One (#69; 5.6%) had previously been found to have BRCA2 c.5213_5216del p.Thr1738Ilefs*2 in combination with NF1 c.6792C>G p.Ala2253_Lys2286del.35 This case was not excluded as, first, the penetrance of pathogenic variants in BRCA2 as BC at age 38 is <20%, and so it could not be assumed that this was the sole reason for this patient’s BC, and, second, to exclude it might risk not finding an important association given the rarity of NF1-BC cases.36
Age of onset
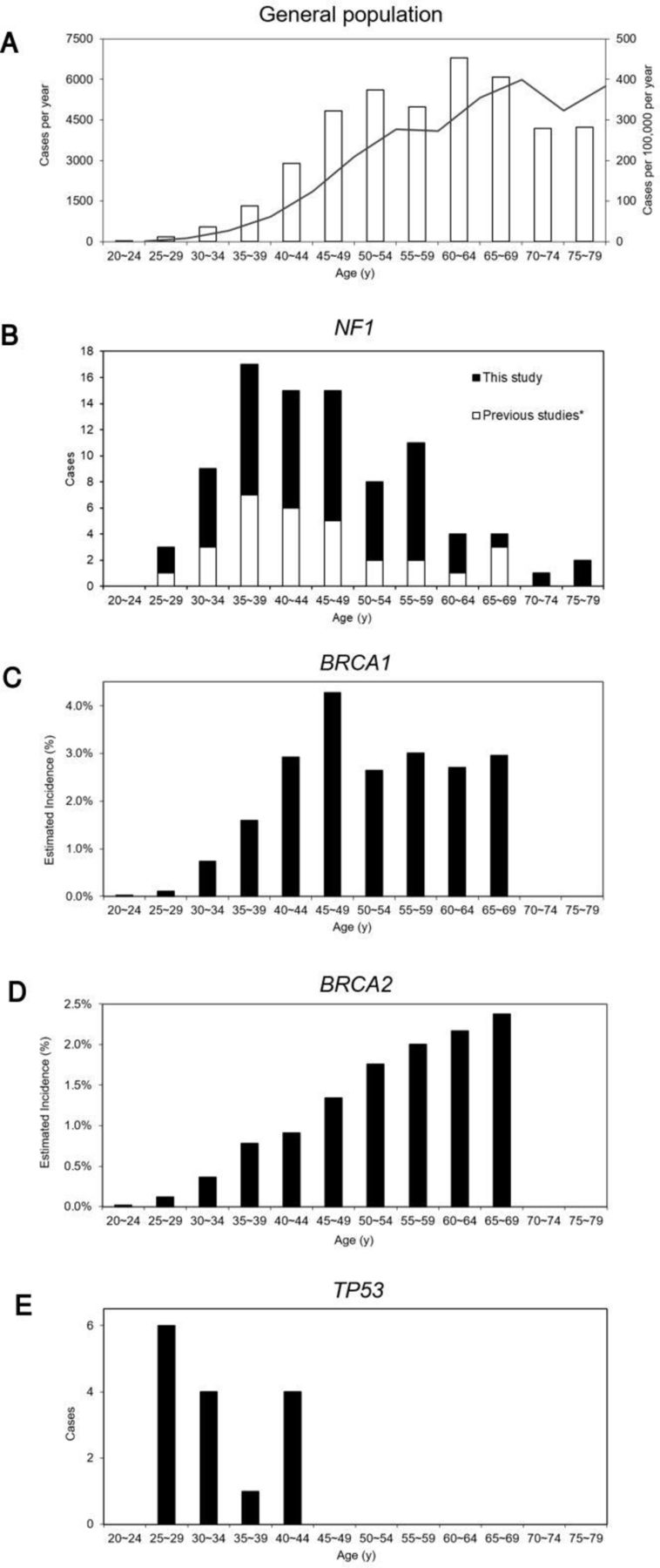
The age of onset of the first or only breast cancer diagnosis was known in 59 (75.6%) cases, giving a mean age of onset of 47.1 years (SD 11.2 years) and a median of 46.0 years, with 37 (62.7%) occurring <50 years. The distribution of ages is shown, in combination with other reports of NF1-BC, in comparison to the UK general population and cases with constitutional BRCA1, BRCA2 and TP53 mutations (figure 1).8 10 11 14 30–32 37
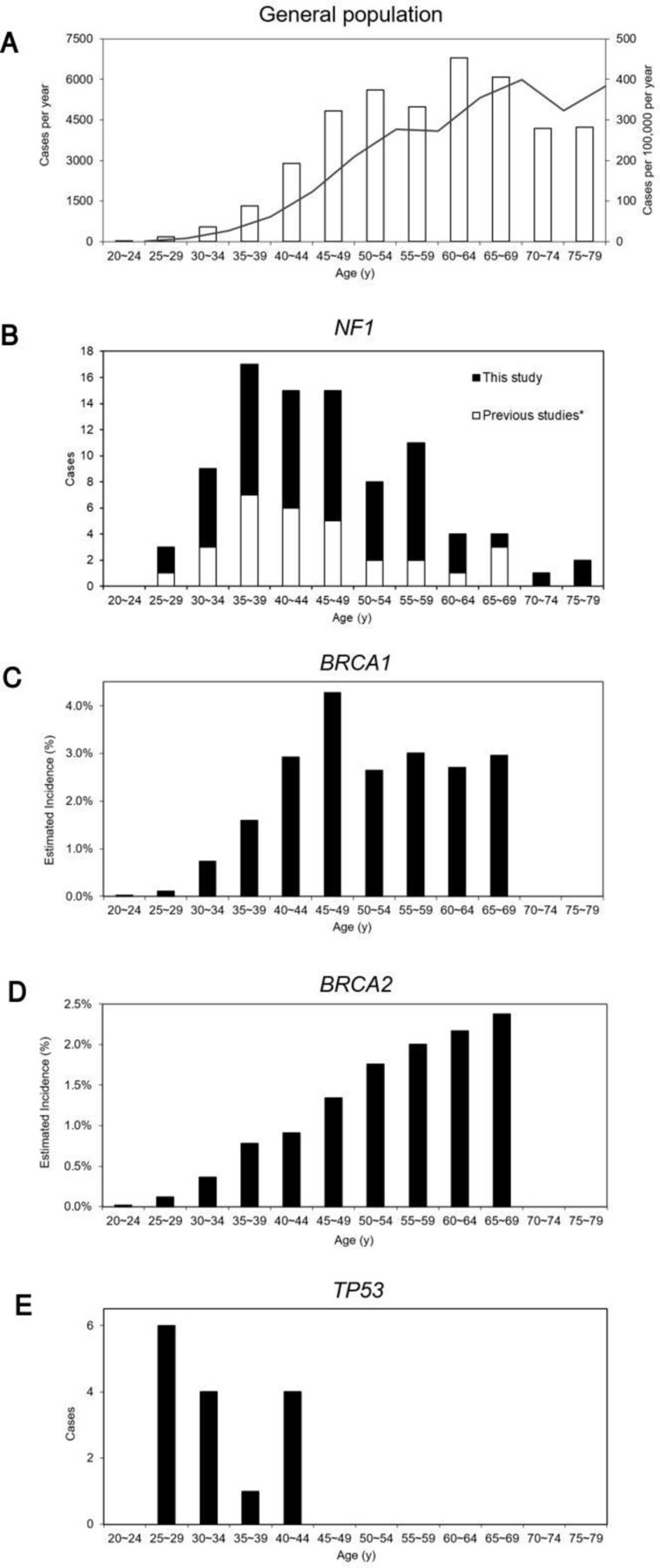
The age distribution of female breast cancer in the general population and those with constitutional NF1, BRCA1, BRCA2 and TP53 mutations. (A) General population (UK)32: □, cases per year; ■, solid line shows cases per 100 000 per year; (B) NF1: ■, this study; □, previous studies8 10 11 14 37; (C) BRCA1 30; (D) BRCA2 30; (E) TP53.31
Constitutional mutation type
Comparison of the seven mutation types: NS, FS, SS, MS, ID, LD and OT by pairwise Fisher’s exact tests showed that the absence of LD in the NF1-BC set was significant (p=0.014) (see online supplementary table 2Sa; online supplementary figure 1S). Applying the Benjamini-Hochberg adjustment procedure for multiple estimates showed that the absence of LD remain significant and the higher proportion of both NS and MS mutations very closely approached significance with borderline adjusted p values of 0.254 and a false discovery rate of 0.25 (see online supplementary table 2Sa; online supplementary figure 1S)38
Supplemental material
Regarding the age of onset of BC in relation to mutation type, it was noted that of the 11 MS cases with known age of onset, 10 occurred at <50 years (p=0.041; Fisher’s exact test). Hence, the analysis was repeated for the 37 cases with BC aged <50 years (see online supplementary table 2Sb). This confirmed the preponderance of MS in younger cases (p=0.009), while no excess of NS is seen. However, in cases aged >49 years no effects can be seen, which is probably a function of the diminished power due to only having 22 cases (see online supplementary table 2Sc).
Constitutional mutation site
Overall
NF1-BC mutations showed a similar overall distribution along the whole coding region as the LOVD, although with a modest under-representation between nucleotides 2001–2400 and 6801–7200 (see online supplementary figure 2S).
Supplemental material
Specific domains
There were no significant differences in the distribution of mutations in the domains CSRD, Tub, GRD, Sec14, PH and Syn compared with the reference LOVD set (see online supplementary table 3S). Although 6 (42.8%) of the 14 MS were observed between codons 709 and 847, this was not a significant concentration in the CSRD domain (p=0.402; Fisher’s exact test) (see online supplementary table 3Sb). Ages of onset of BC in the five females with CSRD MS mutations (#28–#32) were between 29 and 48 years (mean 39.4 years); the sixth case (#33) being a male with BC at age 73.7 years.
Individual constitutional mutations
Of the 78 NF1-BC cases, 63 (87.5%) had truncating or nulling mutations, comprising 31 NS, 19 FS and 13 SS mutations. The remaining 15 cases (7.6%) comprised 14 MS and 1 ID (which as it only involved two amino acid residues, p.Ile1658_Tyr1659del, was included in the MS group for overall comparisons). No cases were observed with OT or LD (eg, microdeletions and intragenic CNV).
It was noted that six pairs of cases shared the same putative protein changes: #15 and #16 with p.Arg440*, #17 and #18 with p.Tyr489*, #32 and #33 with p.Leu847Pro, #52 and #53 with p.Arg1513*, #55 and #56 with p.Tyr1614* and #68 and #69 with p.Ala2253_Lys2286del, plus a trio of cases (#74–#76) with p.Arg2616*, all unrelated. Furthermore, the two pairs of cases with p.Tyr1614* and p.Ala2253_Lys2286del were seen to be caused by different mutations in each pair: c.4841_4842insAAT and c.4842T>G, and c.6792C>A and c.6792C>G, respectively. In addition, 21 (30%) of the 70 different protein changes were not observed in the LOVD. These findings together suggested that the observed frequencies of the mutations in the NF1-BC set were not random. Naturally, common mutations would be expected more often than others, so this was allowed for by calculating the probability of observing each individual mutation in the NF1-BC set given its frequency in the LOVD and plotting the Fisher’s exact p values ranked by probability (Np ) vs 1–p, as described (figure 2) (see online supplementary table 4S).34 The resultant plot of the larger values of p thus gave the straight line with a slope estimating the number of true null hypotheses (T 0), in this instance 20, corresponding to uncorrected p values of ≤0.09 and giving a Benjamini-Hochberg adjusted p value threshold of 0.125 (figure 2).33 34 Hence, 52 of the 70 different mutations observed in the NF1-BC set (74.3%) can be accepted as true associations, that is, related to the occurrence of BC in NF1. The HRs associated with these 52 mutations vary from 83 (95% CI 10.9 to 641) for p.Tyr1614* (p=0.0012; Fisher’s exact test) to 5.3 (95% CI 1.4 to 19.9) for p.Arg440* (p=0.090; Fisher’s exact test) (see online supplementary table 4Sa).

P value plot of the mutations in neurofibromatosis type 1-breast cancer, as ranked Fisher’s exact p values (Np ) vs 1–p. Solid points (●)=individual probabilities (Fisher’s exact test) with the vertical dotted line indicating p=0.05; open points (○)=Benjamini-Hochberg adjusted probabilities, with the solid line intercept indicating T 0, the number of probable true associations and the vertical dashed line showing the resultant Benjamini-Hochberg adjusted p value threshold of 0.125.
Individual mutations and age of onset
Given the younger age of BC onset in MS cases, we then looked further at age of onset. In those with mutations with Benjamini-Hochberg adjusted p<0.125 (ie, significantly enriched), 28/43 (65%) had an age of onset <50 years (mean 45.6 years; median 44.8 years; range 29–76 years), while in those with p>0.125, 9/17 (53%) were <50 years (mean 50.8 years; median 49 years; range 29.8–76 years), consistent with predisposition to BC by the significantly enriched NF1 mutations (see online supplementary table 4Sa; t-test of means: two-sample assuming equal variances: p(T≤t) one-tail=0.052). However, it was noted that the mean age of onset in those without significantly enriched mutations (50.8 years) was also low. To explore this age effect, we repeated the analysis on the 37 cases (with 35 different variants) whose age of onset was <50 years (see online supplementary table 4Sb). This shows that 27 (73%) of these variants can be accepted as true associations (p<0.05/Benjamini-Hochberg adjusted <0.125), and moreover that the HRs for these now vary from 299 (95% CI 119 to 7457) for the nine highest ranked variants (p=0.010; Fisher’s exact test) to 1.8 (95% CI 6.6 to 24.7) for p.Ala2253_Lys2286del (p=0.061; Fisher’s exact test).
Discussion
This large 5-year international study, the first of its kind, has examined possible correlations of NF1 genotype with BC in 78 unrelated patients with NF1-BC. Although there are no simple gross relationships with mutation position, our findings indicate that the constitutional NF1 mutations seen in patients with NF1-BC are not random:
A complete absence of large or whole gene deletions in patients with NF1-BC, plus evidence of an overall higher proportion of both NS and MS mutations. While patients with NF1 with large or whole gene deletions (‘microdeletions’) are known to have more severe disease and reduced longevity, and so might be less likely to develop BC, this would have to be of an extreme degree to have significantly biased our findings.
A majority (74.3%; 52/70) of the protein changes observed in NF1-BC cases are significantly enriched, that is, are more frequent than expected, being above the T 0 intercept on the p value plot (figure 2). Moreover, 30% (21/70) have not previously been reported to the NF1 LOVD.
Two pairs of patients with NF1-BC share the same predicted effects on neurofibromin, but have different mutations at the DNA level (p.Tyr1614* caused by c.4841_4842insAAT and c.4842T>G, and p.Ala2253_Lys2286del caused by c.6792C>A and c.6792C>G). This is reinforced by three recent studies: one of 14 patients with NF1-BC in which a pair of cases was found that share a deep intronic splicing mutation (c.1260+1604A>G), and two others reporting NF1-BC cases with NF1 c.2540T>C p.(Leu847Pro).22 39 40
The age of onset in those with significantly enriched mutations (45.6 years) is lower than in those without (50.8 years; p=0.052) and 91% (10/11) of MS cases with known age of BC onset occurred <50 years. Although non-significant, the occurrence of 6/14 (42.8%) MS mutations within the CSRD is, again, consistent with the other two reports of NF1-BC with NF1 p.(Leu847Pro).22 39
Regarding age of onset, any increased risk of BC attributable to certain NF1 mutations would be expected to manifest more among younger-onset NF1-BC cases. As the processes leading to sporadic BC are likely to apply equally to patients with NF1 as in the general population and the SIR for NF1-BC at ages ≥50 years is approximately 2, no >50% of BCs in this age group would be linked to particular mutations and the rest would have developed BC even if they had no NF1 mutation. However, among women with NF1-BC <50 years, the proportion of cancers associated with the constitutional NF1 mutation is greater (65% vs 53%). Notwithstanding this, those without significantly enriched mutations also show a low mean age at diagnosis (50.8 years), suggesting that their NF1 mutations may also somehow lead to increased susceptibility.
This whole body of evidence is consistent with certain constitutional mutation types, and indeed probably certain specific mutations in NF1 conferring greater or lesser risks of BC, in the absence of high-penetrance mutations in other BC genes. However, whether on their own or as instances of multiple inherited neoplasia alleles syndrome (MINAS), they provide insight into possible mechanisms.
Potential sources of bias
Inherently, there are biases in the ascertainment of all clinical data. The lack of patients with microdeletions (leading to possible reduced longevity) has already been mentioned. However, while patients with NF1 have on average an 8%–15.8% lifetime risk of an MPNST, and patients with microdeletions have a 16%–26% risk, we can find no quantitative data in the literature on the precise longevity of microdeletion patients. Nonetheless, an approximation can be made by modelling. With a 26% risk of MPNST in patients with NF1 with LD, then, of the 204 LD cases in the LOVD set, 53 would get MPNST (and it is assumed die), and 151 would live without MPNST surviving long enough to be at risk for BC. Excluding the 53 MPNST cases, there would be 3379 cases in the LOVD set, of which 151 had LD. Comparing this with zero LD in the 78 NF1-BC cases, the Fisher’s exact test still yields p=0.049. And this is a conservative estimate, as the risk of MPNST among patients with LD is 16%–26% and patients with other types of mutations can also get MPNST. Thus, the MPNST risk in patients with LD is not sufficient to explain the lack of LD in the current material, although the reduced longevity of patients with LD may cause minor bias.16 41–45 So, until such time as the longevity of LD patients is precisely described, ideally in a prospective study, the conclusion must stand that LD may be associated with a significantly reduced HR for BC.
Patients with NF1 with BC may have been more likely to be offered gene testing, but this effect likely occurs in any patient with NF1 with a more severe or unusual clinical feature and short of a fully prospective randomised trial (probably unfeasible in the setting of a rare condition) is difficult to control for, although by consideration of gene testing dates in relation to the date of BC diagnosis this may be possible in the future. Although we generally excluded mutations of debateable pathogenicity reported to the LOVD, by selecting only those of classes 4 or 5, there may be some bias in the reporting of mutations to the LOVD, in that they may more often be those which are easier to interpret as pathogenic. However, there is no reason to suspect this applies more or less to patients with NF1 with BC.
For patients with BC onset <50 years (more than half of this cohort, where age of onset was known), less than half had been screened for germline BRCA1/2 or TP53 mutations. Hence, undetected high penetrance constitutional mutations, especially in the six subjects with multiple BCs, might have introduced bias. This is difficult to control for in a largely retrospective study in which comprehensive genetic testing has not been carried out, and is one reason for recommending gene panel testing in patients with NF1-BC. However, even if some of these multiple BC cases are due to mutations in other genes, the majority are less likely to be, and in any event they would provide more evidence for MINAS as a factor in NF1-BC.35
Reasons for the relationship of NF1-BC with mutation type
Clearly, NF1 mutation type is related to the risk of NF1-BC, but the effect is subtle. The magnitude of the effect due to NS or MS mutations is only sufficient in itself to explain less than 50% of cases, but it does demand explanation. In particular, why might some truncating mutations due to NS, but not FS or SS mutations, raise the risk of BC? How might this then relate to the findings regarding MS mutations, that is, of restricted types and possible concentration within a specific domain, and the fact that no patients with NF1-BC have been observed to have whole or even partial gene deletions?
Large constitutional deletions of tumour suppressor genes, akin to loss of heterozygosity as a somatic event, cause loss of normal protein function. Missense or NS mutations can theoretically cause a change or gain in function, whereas FS and SS mutations mostly result in loss of function as they strongly stimulate nonsense-mediated decay (NMD) of mRNA.46 Hence, this selective excess of MS and NS mutations suggests that some NS mutations confer a gain of function that may be involved in NF1-BC risk. While NS mutations often stimulate NMD they vary in the extent to which they do this. It is well known that NS mutations in the most 3′ exon and within 50~55 bp of the 3′ splice site of the penultimate exon of a gene do not stimulate NMD, but recent work has shown the process to be much more complex, involving 38 parameters including the ‘last exon’ rule.47 Thus, NS mutations that do not fully stimulate NMD may result in the expression of truncated protein with the potential for a gain of function effect on cellular function. This would also fit with MS mutations being restricted in type and site in NF1-BC, presumably because only certain MS mutations confer the specific changes or gains of function which BC cells find advantageous.
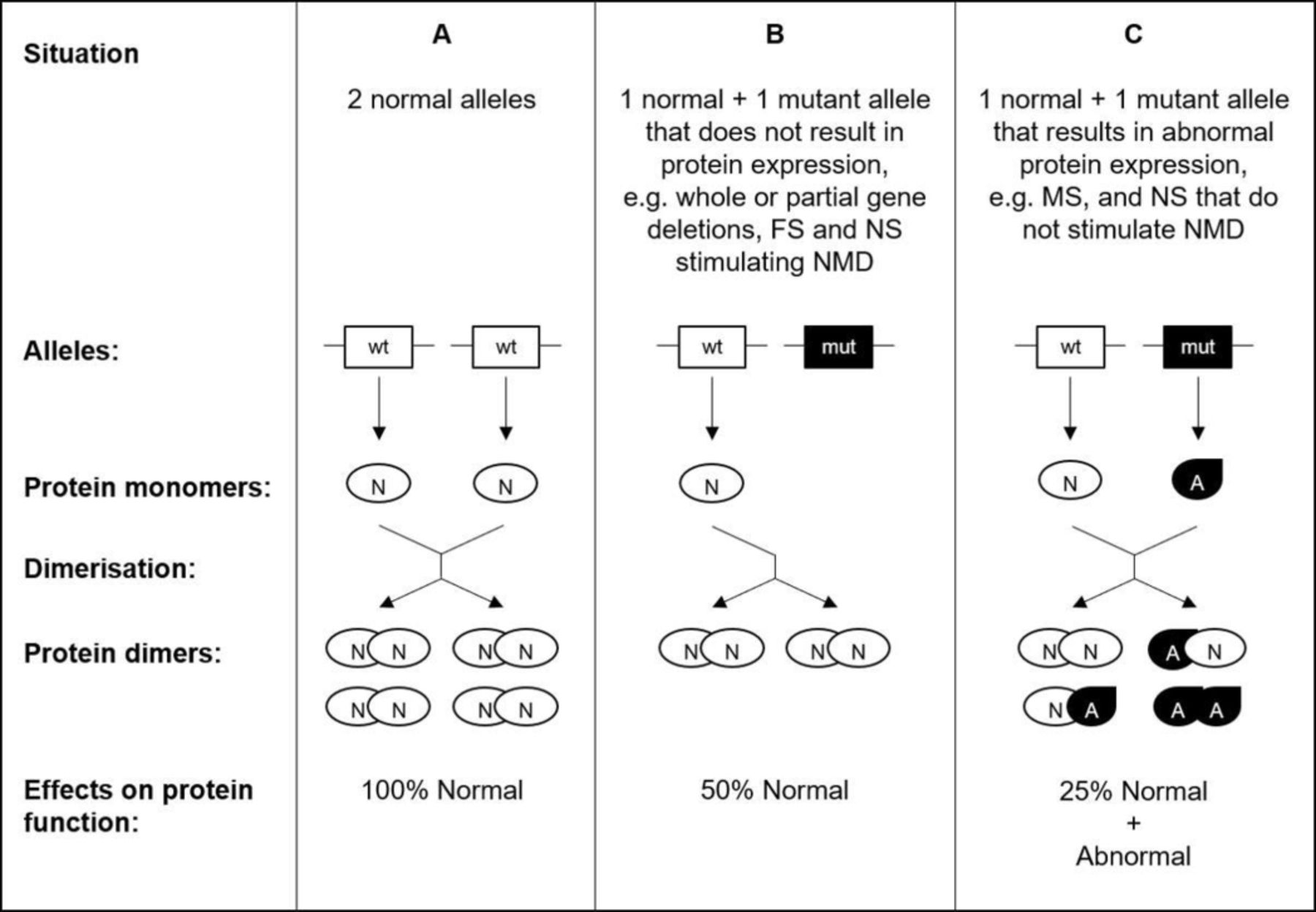
As to the molecular mechanism or mechanisms involved, our genetic findings support the emerging biochemical evidence that neurofibromin acts within the cell as a dimer, which would provide an elegant explanation for our observations.48 49 Mutations causing loss of normal protein expression, such as whole or partial gene deletions, FS and NS stimulating NMD, would result in a simple halving of neurofibromin activity. However, mutations causing expression of abnormal protein, such as MS and NS that do not stimulate NMD, would result in a greater reduction in normal function, and may have abnormal function (figure 3). However, it is accepted that this may be more complex in the case of FS variants, some of which may allow full-length mRNA to be produced, sometimes including a MS. Also, we did not differentiate between FS that generate in-frame and out-of-frame message, as the precise effect of any given FS mutation can be complicated, may involve allele-specific changes in expression levels, and differ according to the assays used. Of course, any one variant might confer gain of normal function and/or loss of other function/s, perhaps by acting dominant-negatively. But, gain of tumour-suppressor function may be by downregulating another function, perhaps by upregulating a third, for example, APC, β-catenin and c-Myc, and it is unlikely that all variants will act in the same way, which is supported by the recent finding that expression patterns of neurofibromin in NF1-BC are not consistent.50 51 Nonetheless, NF1 may thus be similar in this respect to familial adenomatous polyposis, where the tumour suppressor protein APC functions as a dimer, and, moreover, provides an explanation for that disease’s variable severity through the resultant constraint by a patient’s constitutional mutation on which somatic mutations confer a selectable advantage.52

{kind=link}
![[jmedgenet-2018-105599supp002.jpg]](https://jmg.bmj.com/content/jmedgenet/56/4/209/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
![[jmedgenet-2018-105599supp003.jpg]](https://jmg.bmj.com/content/jmedgenet/56/4/209/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
{kind=link}
A diagram schematically illustrating the potential effects of mutations on proteins that function as dimers (A). Mutations that result in non-expression of normal protein from one allele (B) simply result in a reduction of normal function by half. Such mutations include, eg, whole or partial gene deletions, frameshift (FS) and nonsense (NS) stimulating nonsense-mediated decay (NMD). However, with mutations such as missense (MS) and NS that do not stimulate NMD (C), the result is the expression of abnormal protein. Such abnormal monomers result in the formation of normal-abnormal heterodimers and abnormal homodimers, the effect of which is twofold: to reduce normal function by three-quarters as well as confer a gain of abnormal function. This assumes that parameters such as expression levels and binding coefficients are not altered and is necessarily a simplification.
The recent finding that BC stands out among cancer types in being the only one in which amplification of NF1 is seen frequently as a somatic event also supports the contention that gain or change of function is important as a selectable advantage in BC tumourigenesis.25 39 53 However, ERBB2 amplification is a known driver of BC, and so the amplification of NF1 in sporadic BCs might simply be a function of the two genes being linked on chromosome 17. We therefore analysed data from five studies in the cBioPortal for Cancer Genomics, including the Metabric dataset, and while in 541 cases of BC with changes in NF1 or ERBB2 the majority (376, 69.5%) had ERBB2 amplification, only 9.6% (36/376) had NF1 amplification and four (1.1%) had homozygous deletions of NF1 (see online supplementary table 5S).54 In contrast, 36/44 tumours with NF1 amplification also had ERBB2 amplification. Clearly, although most BC with NF1 amplification also have ERBB2 amplification, most BC with ERBB2 amplification do not have NF1 amplification. Therefore, on some occasions, at least, NF1 gene amplification is likely an independent selectable advantage, and while NF1 amplification is commonly accompanied by ERBB2 amplification this does not necessarily imply the NF1 amplification is a mere passenger.
Clinical utility of our findings
If it were possible to identify precisely a subset of NF1 mutations that confer a raised risk of NF1-BC then that would be clinically useful. While we have identified a group of 52 mutations as probably significantly associated with NF1-BC, it is difficult to single any one out as conferring a clinically significant increased risk of BC, although their individual HRs are all between 5.3 and 148 (95% CI 1.4 to 3669), reinforced by many showing a greater effect <50 years, which the reader may consider sufficient to act on. But, that said, the evidence from this study is adding to others indicating a greater risk of malignancy in general associated with MS mutations involving codon 847.22
In contrast, the low risk of BC associated with large NF1 deletions may be more clinically useful, although that it too needs to be confirmed in other datasets. Given that we ascertained no cases with LD in 78 patients with NF1-BC and 204 cases with LD are reported on the LOVD (5.9% of 3432), then the HR is 0.10 (95% CI 0.006 to 1.63; p=0.014, Fisher’s exact test). It needs to be borne in mind that the upper CI exceeds 1 due to the necessity to add 0.5 to all numbers in the calculation, because one number equals zero, and also that the distribution of the CI around an HR is heavily skewed to the lower CI. In contrast, Fisher’s calculation of the exact probability does not need this internal addition. It is therefore curious that such microdeletion cases appear less likely to develop BC considering NF1 microdeletion cases have a twofold increased risk of developing MPNSTs.16 41–45 As SUZ12 potentiates the effects of NF1 mutations, however, by amplifying Ras-driven transcription through effects on chromatin, and SUZ12 is only 0.58 Mb telomeric of NF1, it is thus frequently lost in tumours from microdeletion cases. Hence, this may indicate that a reduction in SUZ12 may protect against NF1-BC.16 55
While only 1 out of the 18 NF1-BC cases (5.6%) tested for mutations in other genes had a mutation (in BRCA2), nonetheless, because BRCA1 is linked to NF1 and cases with both NF1 and BRCA1 mutations have been described, then it would be logical and reasonable to test NF1-BC cases for mutations in other BC-predisposing genes, as possible cases of MINAS.10 35 56 Certainly, it would be unreasonable to assume that a case of NF1-BC was always solely due to the constitutional NF1 mutation. This, therefore, argues for more extensive genetic testing being offered to those with NF1, for example, in the form of gene panels, and highlights the benefit of such an approach. Vice versa, what value is there in testing NF1 in all BC cases? One study has reported finding pathogenic or likely pathogenic NF1 variants in 153 of 108 883 (0.1%) cancer gene panels, of which 4 (3%) were reported not to have any clinical features of NF1.57 So, for reasons such as mosaicism, phenotype restriction or clinical inattentiveness, the occasional patient may not be diagnosed clinically with NF1 before they are molecularly diagnosed. However, the yield of otherwise undiagnosed NF1 cases from cancer gene panels is exceedingly low at approximately 0.004%. So, whether it is worth including NF1 on such panels in the absence of clinical features is perhaps a question more for health economics.
Summary
We have shown that:
The type of heritable NF1 mutation is a determinant of BC risk in NF1. However, this effect does not fully explain the phenomenon of BC in NF1.
Particular point mutations, including MS mutations in the CSRD, may specifically confer an increased risk of BC, and more so at younger age.
Patients with NF1 with large or partial gene deletions may have little or perhaps no increased risk of BC.
The mechanism of this effect may relate to the degree to which individual mutations result in a change of neurofibromin function and exactly what function/s.
There is a reasonable chance a patient with NF1-BC may harbour a mutation in another BC predisposing gene, so testing for constitutional mutations in genes other than NF1 should be offered.
As BC risk in NF1 may be a function of specific mutations conferring gain of function, then, in order to address this hypothesis, and confirm our other findings, a prospective NF1-BC clinical database should be established, combined with comprehensive constitutional and tumour genomic analysis. As the cases in this study not already listed will be deposited in the LOVD, then this could be achieved within the NF1 LOVD by the simple addition of the dates on which patients were diagnosed with an NF1 mutation, and the dates on which tumours are diagnosed. We therefore strongly recommend that all clinicians who care for patients with NF1 submit their data to the LOVD.
Abstract translation
This web-only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.Acknowledgments
The authors would like to thank Professor Ludwine M Messiaen for sharing data and advice, as well as Sheila Palmer-Smith, John Watkins and Professors Ros Eeles and David N Cooper for advice and helpful discussions. The authors would like to acknowledge the data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
References
Footnotes
Contributors V-FM and MU had the original idea for the study, IMF and MU designed it. IMF sought institutional approval, compiled, checked and analysed the data, managed the study database, computed the results and wrote the initial draft of the manuscript including text, tables and figures. RAK, JP and IMF worked on the statistical analyses. RvM compiles, checks and curates the online NF1 database (LOVD); RvM and IMF shared appropriate data. DGE, JP and MU reviewed the initial draft of the manuscript. IMF, V-FM, RvM, RAK, SA, DB, SB-S, AC, HC, DME, SF, HLD, CL, MTR, AJS, PS, AV, YSY, OZ, JP, DGE, PW and MU all contributed data, critically reviewed study design, interpretation of mutations and results and commented on and approved the final manuscript. IMF submitted the study and is responsible as guarantor for overall content.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests Dr IMF reports support from St Vincent’s University Hospital, Dublin, Impact Genetics, Bowmanville, Ontario, Canada and Ambry Genetics, Aliso Viejo, California, USA, and Professor GDE reports support from AstraZeneca, both for travel, both outside the submitted work. Professor MU thanks the Ian Owen Fund for support.
Ethics approval Cardiff and Vale University Health Board (13/DHD/5637).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement NF1 gene variant data not already listed on the public LOVD at https://databases.lovd.nl/shared/genes/NF1 will be deposited on the LOVD. We welcome submission of further NF1 breast cancer cases to the LOVD to extend the study, and request that submitters contact the corresponding author. Within the limitations of the original retrospective study, as stated in the paper, there are some extra clinical data which can be made available to those contributing more cases.
Correction notice The article has been corrected since it was published online first. The name of author D Gareth Evans has been corrected.
Patient consent for publication Cardiff and Vale University Health Board approved the study as not requiring specific ethical approval, because it only used anonymous data collected during routine patient care, and hence individual patient consent was not necessary (13/DHD/5637).