Article Text

Abstract
Background Four genetic causes of isolated congenital central hypothyroidism (CeH) have been identified, but many cases remain unexplained. We hypothesised the existence of other genetic causes of CeH with a Mendelian inheritance pattern.
Methods We performed exome sequencing in two families with unexplained isolated CeH and subsequently Sanger sequenced unrelated idiopathic CeH cases. We performed clinical and biochemical characterisation of the probands and carriers identified by family screening. We investigated IRS4 mRNA expression in human hypothalamus and pituitary tissue, and measured serum thyroid hormones and Trh and Tshb mRNA expression in hypothalamus and pituitary tissue of Irs4 knockout mice.
Results We found mutations in the insulin receptor substrate 4 (IRS4) gene in two pairs of brothers with CeH (one nonsense, one frameshift). Sequencing of IRS4 in 12 unrelated CeH cases negative for variants in known genes yielded three frameshift mutations (two novel) in three patients and one male sibling. All male carriers (n=8) had CeH with plasma free thyroxine concentrations below the reference interval. MRI of the hypothalamus and pituitary showed no structural abnormalities (n=12). 24-hour thyroid-stimulating hormone (TSH) secretion profiles in two adult male patients showed decreased basal, pulsatile and total TSH secretion. IRS4 mRNA was expressed in human hypothalamic nuclei, including the paraventricular nucleus, and in the pituitary gland. Female knockout mice showed decreased pituitary Tshb mRNA levels but had unchanged serum thyroid hormone concentrations.
Conclusions Mutations in IRS4 are associated with isolated CeH in male carriers. As IRS4 is involved in leptin signalling, the phenotype may be related to disrupted leptin signalling.
- IRS4
- central hypothyroidism
- leptin
- HPT Axis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Central hypothyroidism (CeH) is characterised by thyroid hormone deficiency due to insufficient production of thyroid-stimulating hormone (TSH).1 Congenital CeH is often part of multiple pituitary hormone deficiency, but isolated TSH deficiency occurs in 20%–25% of cases. Known genetic causes of congenital isolated CeH include mutations in TSHB, TRHR, IGSF1 and TBL1X. 2–5 However, many cases remain unsolved.
In this study, we used ‘whole’ exome sequencing (WES) to identify the genetic cause of isolated CeH in two unrelated families. We identified mutations in insulin receptor substrate 4 (IRS4) in both families. Sanger sequencing in other cases of CeH identified IRS4 mutations in three families. The IRS family acts as interface between tyrosine kinase receptors, including the insulin, leptin and insulin-like growth factor 1 (IGF-1) receptors, and multiple intracellular signalling pathways.6 Although the precise function of IRS4 is unclear, its discrete expression in rat hypothalamic nuclei suggests a specialised neuroendocrine function(s).7
Methods
Patient acquisition
In contrast to TSH-based neonatal congenital hypothyroidism (CH) screening programmes, the Dutch thyroxine (T4)+TSH+T4 binding globulin (TBG)-based screening effectively detects primary CH and congenital CeH.8 9 Many of these children are treated by the Department of Paediatric Endocrinology of the Academic Medical Center, University of Amsterdam.10 We studied two pairs of brothers: 18-year-old A.III.4 and his 14-year-old brother A.III.5 (figure 1A), and 12-year-old B.III.4 and his 22-year-old maternal half-brother B.III.3 (figure 1B). The first three were detected by neonatal screening and were diagnosed with congenital CeH at the age of 2 weeks. B.III.3, who was born before optimisation of the Dutch neonatal screening programme for CeH, had normal screening results and was diagnosed after presenting with short stature and delayed tooth eruption at age 12 years (table 1). In all four, the diagnosis CeH was supported by thyrotropin-releasing hormone (TRH) stimulation testing (online supplemental table 1), while additional endocrine testing demonstrated normal functioning of the other hypothalamic–pituitary axes. Levothyroxine (LT4) treatment was started in all. While A.III.5, B.III.3 and B.III.4 have grown and developed normally, A.III.4 had a delayed pubertal growth. Testosterone-primed growth hormone (GH) stimulation tests were normal. With spontaneous progression of puberty, his growth improved, resulting in a normal adult height (online supplemental table 2). All four patients tested negative for mutations in TSHB, TRHR, IGSF1, and TBL1X.
Supplementary file 1
Characteristics of probands and relatives with IRS4 mutations

Pedigrees of five families with IRS4 mutations. Probands are indicated by an arrow; small horizontal lines indicate that DNA sequence analysis was performed. Black filled symbols represent mutation carrying individuals. (A) Pedigree of family A, (B) family B, (C) family C, (D) family D and (E) family E.
Genetic analyses
Genomic DNA isolation was performed as described previously.4 WES and variant calling were conducted using BWA-MEM (0.7.5), GenomeAnalysisTK-2.8–1-g932cd3a, Cartagenia 3.0. and the SeqCap EZ Human Exome Library v3.0 kit (NimbleGen; Roche, Madison, Wisconsin, USA) on a HiSeq2000 (Illumina, San Diego, California, USA). Variants were filtered and analysed as described previously.5 Candidate variants were confirmed by Sanger sequencing. WES was performed in the four probands; Sanger sequencing was performed in all their available first-degree and second-degree relatives and in other patients with idiopathic isolated CeH. Written informed consent was obtained in all cases.
Phenotyping
Clinical studies
Mutation carriers underwent assessment of growth and development, biochemical evaluation of hypothalamus–pituitary axes and glucose homeostasis, thyroid ultrasound and pituitary MRI. Two patients (A.III.4 and B.III.3) underwent 24-hour blood sampling to assess TSH secretion, as described earlier.11 Testicular ultrasound was performed in all male mutation carriers aged 12 years or older. Mutation carriers additionally underwent hearing assessment by pure tone audiometry (PTA) or otoacoustic emission testing. PTA was performed in a soundproof booth, using a manual audiometer (Madsen Electronics, Taastrup, Denmark) with TDH-39 headphones, calibrated according to ISO-389–1, with adequate masking.12
Endocrine measurements
Plasma free T4 (FT4) and prolactin concentrations were measured by fluoroimmunoassay using the Delfia 1232 Fluorometer (Wallac, Turku, Finland), while total T4, triiodothyronine (T3) and reverse T3 (rT3) were measured by an inhouse radioimmunoassay (RIA).13 Plasma TSH, luteinizing hormone (LH), follicle-stimulating hormone (FSH) and 17β-oestradiol were measured by electrochemiluminescence assay using the Roche cobas e602 (Roche Diagnostics, Mannheim, Germany). Plasma TBG and serum thyroglobulin were measured by a RIA (BRAHMS, Thermo-Scientific Hennigsdorf, Germany).
Plasma testosterone was measured by an inhouse method using the Acquity ultra-performance liquid chromatography-tandem mass spectrometry system (Waters, Milford, Massachusetts, USA). Serum sex hormone-binding globulin (SHBG) was measured with immunoluminometric assay (ILMA) using the Architect iSR2000 (Abbott Laboratories, Diagnostics Division, Illinois, USA). Serum IGF-1 and GH was measured with electrochemiluminiscence assay using the Liaison (Diasorin, S.P.A., Saluggia, Italy). Plasma insulin, insulin growth factor binding protein 3, adrenocorticotropic hormone and cortisol were measured by ILMA using the Immulite 2000 (Siemens Medical Solutions, Camberley, UK).
Plasma total cholesterol was measured with enzymatic colorimetric assay using the Roche cobas c502, while triglycerides and glucose were measured with enzymatic colorimetric assay using the Roche cobas c702 (both from Roche Diagnostics). Plasma leptin concentrations were measured with a RIA from Millipore (Billerica, Massachusetts, USA).
mRNA expression in human tissue samples
Three unfixed, frozen (−80°C) human hypothalami and pituitaries were obtained from the Netherlands Brain Bank (NBB) and used for mRNA expression. Permission for brain autopsy and for the use of human brain material and clinical information for research purposes had been obtained according to the protocols of the NBB. Serial coronal 50 µm sections were cut from unfixed frozen hypothalami on a cryostat. We collected all consecutive sections from the level of the lamina terminalis to the mammillary bodies as previously described.14 RNA was extracted from the supraoptic, paraventricular and infundibular nuclei, and lateral hypothalamic area and from the homogenised pituitaries using TriReagent (Sigma) per the manufacturer’s instructions, followed by DNase treatment (Qiagen GmbH, Germany). cDNA was synthesised with an Applied Biosystem Kit. From every sample, an RT reaction was performed in order to check for genomic DNA contamination.
PCR
Primers were designed to amplify IRS4 transcript (NM_003604.2, F:5′-CCCCCGGAGAGAGAAGAT-3′, R: 5′-ACTGTAGACTGTAGCGCATCG- 3′, product size 171 bp, annealing temperature 65°C). Real-time PCR was performed using the Lightcycler480 (Roche Molecular Biochemicals, Mannheim, Germany) and SensiFAST SYBR No-ROX Kit (Bioline, London, UK). Melting curve analysis was performed, and product size was determined by DNA gel analysis. All samples contained mRNA as checked by hypoxanthine phosphoribosyl transferase (HPRT) expression (a housekeeping gene).14
Mouse studies
Irs4 knockout (KO) (hemizygous males and homozygous females) and wild-type (WT) (B6129SF2/J) mice (n=6 per group) were bred at Jackson Laboratories (Jax, Bar Harbour, Maine, USA). Serum, brain and pituitaries from mice that are 6–8 weeks old were collected at the Jax facility, frozen and shipped on dry ice. We measured serum thyroid hormones, and Trh and Tshb mRNA expression in brain tissue.
Serum thyroid hormone concentrations
Serum TSH levels were measured using a mouse Pituitary Magnetic Bead panel for TSH (MILLIPLEX), following the instructions of the manufacturer (Merck-Millipore, Darmstadt, Germany) and read on a BioPlex (BioRad). Total T4 and T3 were measured by an inhouse RIA.13 In order to prevent interassay variation, all samples of one experiment were measured within the same assay (intra-assay variation: TSH: 9%–12%, T3: 3.6% and T4: 6.6%).
RNA isolation and qPCR
Total RNA from the pituitary was isolated using TriReagent (Ambion) and the Nucleospin RNA kit (Macherey-Nagel, Duren, Germany). RNA concentrations were determined using the Denovix spectrophotometer (Denovix, Wilmington, Delaware USA). The cDNA synthesis was carried out using equal RNA input and the first strand AMV cDNA synthesis kit (Roche Molecular Systems, Pleasanton, California, USA). Quantitative PCR was performed using the Lightcycler 480 apparatus and the SensiFAST SYBR No-ROX Kit (Bioline) and according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines. Quantification was performed using the LinReg software. Samples with a mean deviation of more than 5% of the mean efficiency value of the assay were excluded. Calculated values were normalised by the geometric mean of three reference gene values (HPRT, EF1a1 and cyclophilin), which were selected to be the most stable among different groups. The primers used were previously described.15 16
In situ hybridisation
Twenty-micrometre brain sections were cut on a cryostat (Leica). Riboprobe synthesis and in situ hybridisation for Trh mRNA were carried out as reported before.15
Statistics
Student’s t-test was used to compare results of patients and controls (SPSS V.23 for Windows). P value of <0.05 was considered significant. For the mouse studies, effects of sex and genotype were assessed by two-way analysis of variance followed by a post hoc test (Tukey’s or Sidak’s) with GraphPad software (GraphPad Prism 7, La Jolla, California, USA).
Results
WES revealed nonsense or frameshift mutations in X-linked IRS4 (transcript accession number NM_003604.2) in the two pairs of brothers. Sanger sequencing in 12 other unrelated patients with congenital isolated CeH revealed three (two novel) frameshift mutations in IRS4 in three patients (table 1, figures 1C–E and 2): two 5-year-old boys (C.III.2 and E.III.4), and a 10-month-old boy (D.III.6). All three had been detected by neonatal screening and were treated with LT4. Off-treatment re-evaluation of the hypothalamus–pituitary–thyroid (HPT) axis in patients C.III.2 and E.III.4 at age 3 years confirmed the initial CeH diagnosis in C.III.2, and LT4 was restarted; although E.III.4 had repeatedly too-low neonatal FT4 concentrations and a blunted TRH test in the neonatal period, re-evaluation showed FT4 concentrations around the lower limit of the reference interval (10.3 pmol/L). Because of clinical signs of hypothyroidism, he was also restarted on LT4. All three boys grew and developed normally. All probands tested negative for mutations in TSHB, TRHR, IGSF1 and TBL1X. The IRS4 mutations were found in 14 relatives (one male; E.III.5). Ten individuals were included in the study, while four women (two adults, two children) declined participation. Three-year-old E.III.5 was diagnosed with CeH during endocrine evaluation for the present study and subsequently started on LT4. In retrospect, he had a low neonatal screening total T4 concentration with low TBG (129 nmol/L; neonatal reference interval: 160–750 nmol/L).17

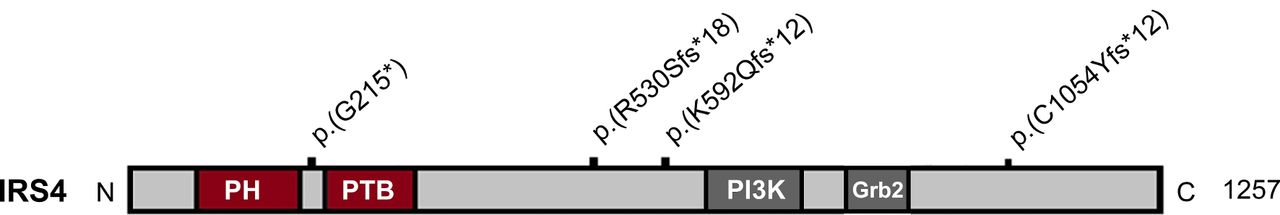
Schematic of IRS4 and the positions of the mutations. PH, pleckstrin homology domain; PTB, phosphotyrosine-binding domain; PI3K, phosphoinositide 3-kinase (region containing multiple PI3K binding motifs); Grb2, growth factor receptor-bound protein-2 (Grb2 binding site). Mutations are indicated with black bars.
Families B and E had the same c.1772dupG mutation (table 1), which was found with a minor allele frequency of 0.47% in the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org; none hemizygotes), but did not occur in GoNL (http://www.nlgenome.nl/), Scripps Wellderly (https://genomics.scripps.edu/browser/) or in 300 house reference samples. The other three mutations (A: c.643G>T; C: c.3161_3165del; D: c.1587_1588insT) were novel.
Clinical characteristics
Endocrine and anthropometric findings
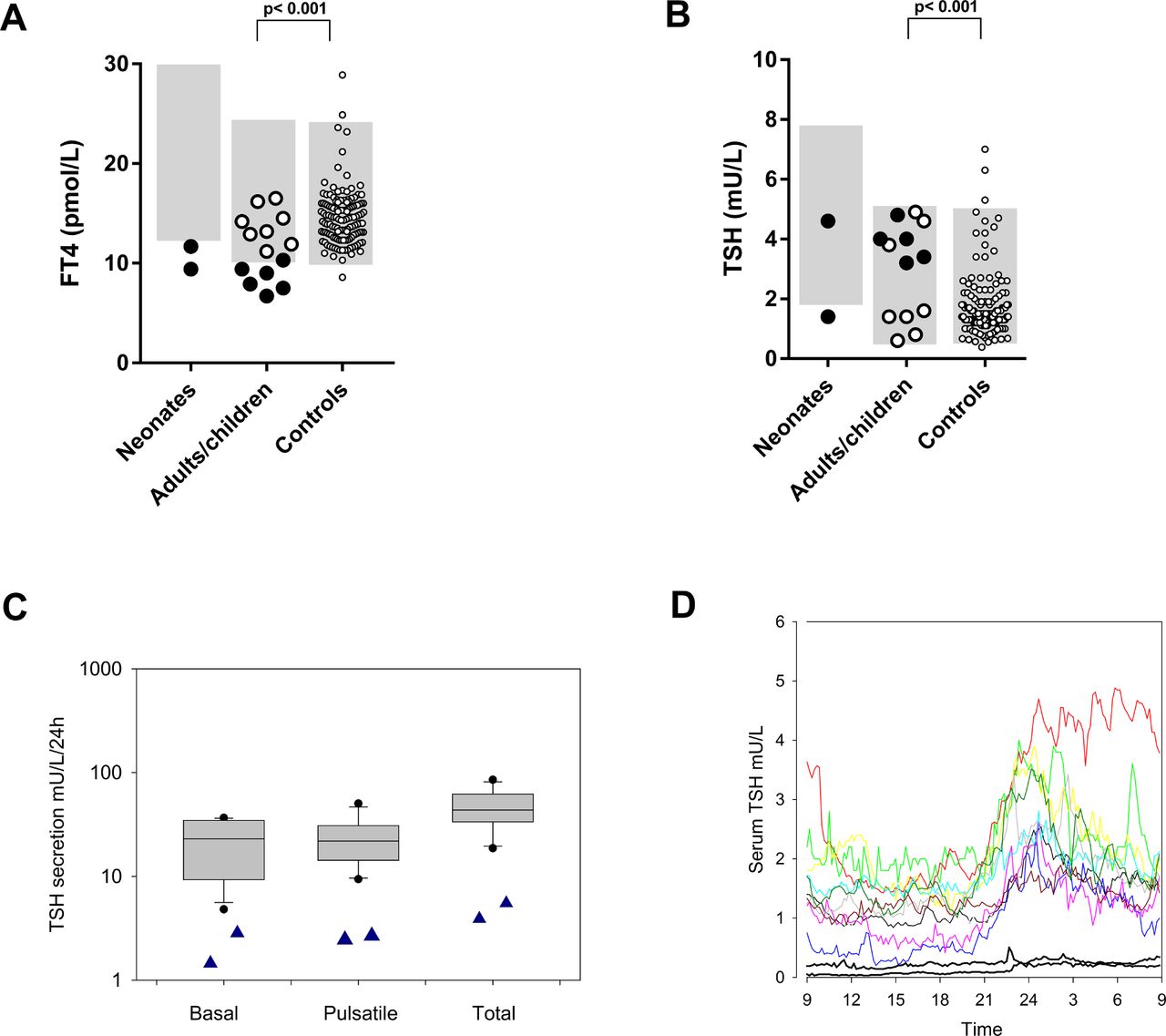
All male IRS4 mutation carriers (n=8) showed biochemical CeH, with FT4 concentrations between 72% and 97.5% of the lower limit of the reference interval (table 1, figure 3A,B). Female heterozygous carriers (n=8) had FT4 concentrations within the lower half of the reference interval. One female carrier had a slightly elevated T3 concentration (A.II.6), while all others had T3 concentrations within the reference interval. T3/T4 ratios were elevated compared with those of euthyroid adult controls.18 TRH testing (performed before LT4 treatment) showed a blunted peak TSH in six of seven males, compatible with CeH (online supplemental table 1).

{kind=link}
{kind=link}
{kind=link}
Plasma FT4 and TSH concentrations. (A) plasma FT4 and (B) TSH concentrations in untreated condition. Neonates: neonates with IRS4 mutations; adults/children: adults and children with IRS4 mutations (black filled symbols: males; white filled symbols: females); controls: adult controls. The grey shaded areas denote the reference intervals. Reference intervals for FT4: 10–23 pmol/L (adults and children) and 12–30 pmol/L (neonates), and TSH: 0.5–5 mU/L (adults and children) and 1.7–7.9 mU/L (neonates). The adult controls (n=136) were recruited in earlier studies for the express purpose of establishing reference intervals and were all healthy subjects, not suspected to have endocrine disorders.5 (C) basal, pulsatile and total TSH secretion of individuals with IRS4 mutations (blue triangles) and healthy controls (box plots). (D) the 24-hour secretion profiles of TSH in individuals with IRS4 mutations (black lines) and healthy controls (coloured lines). FT4, free thyroxine; TSH, thyroid-stimulating hormone.
Mean (fasted) IGF-1 plasma concentration was +1.1 SD score. Serum leptin concentrations were within the age-specific and sex-specific reference intervals in 12 carriers and above the reference interval in four carriers (two infants, two adult females). Height, body mass index (BMI) and biochemical evaluation of other hypothalamus–pituitary axes were unremarkable (online supplemental table 3).
Oral glucose tolerance tests (OGTT) in carriers aged 6 years or older (online supplemental table 4) demonstrated normal glucose concentrations, both fasting and 2 hours after ingestion of a glucose load. Homeostatic model assessment of insulin resistance (HOMA-IR) showed insulin resistance in 3 of 15 individuals (online supplemental table 5). Data on low-density lipoprotein, total cholesterol concentrations and sex hormone-binding globulin concentrations are given in online supplemental tables 3 and 6.
Twenty-four-hour TSH secretion profiles in 2 adult male patients and 11 healthy controls (online supplemental table 7) showed markedly decreased basal, pulsatile and total secretion of TSH in patients, even though their FT4 concentrations were significantly lower (figure 3C,D).
Imaging
Pituitary MRI in 12 carriers showed normal morphology of the hypothalamus and the pituitary. Ultrasonography showed thyroid volumes below the 2.5th percentile in two adult males (both treated with LT4), and well below the 50th percentile in five boys (three treated with LT4) (table 1).19 Thyroid volumes were below the 50th percentile in seven of eight adult women (none treated with LT4).20 In a 1-year-old boy (treated with LT4) and a 6-month-old girl (not treated with LT4), thyroid volumes could not be calculated reliably because of their age. Testicular volume was normal in the four adolescent and adult men.21
Audiometry
Three of 15 carriers (all adults, one male) had hearing thresholds poorer than the age-specific reference interval without clinical manifestations.22
Tissue studies
IRS4 mRNA was expressed in the several human hypothalamic nuclei, including the paraventricular nucleus, and the pituitary gland (online supplemental figure 1).
Mouse studies
Trh mRNA expression in the paraventricular nucleus of the hypothalamus was assessed with in situ hybridisation. No significant differences in hybridisation signal were observed between WT and KO mice, or between male and female mice (online supplemental figure 2A). Tshb mRNA in the pituitary was markedly decreased (p=0.04) in KO females compared with WT females, but no differences were observed in males (online supplemental figure 2B). This difference was not reflected in serum TSH, T4 or T3 concentrations. In both the WT and KO groups, females had lower serum TSH levels than males. Serum T4, but not T3, was higher in WT females compared with males. No differences in either serum TSH, T4 or T3 were observed between KO mice compared with WT mice (online supplemental figure 2C–E).
Discussion
In this study, we identified four mutations (one nonsense and three frameshift) in IRS4 in seven male patients with CeH from five families. Further investigations yielded mutations in 14 relatives, including one male who was subsequently also diagnosed with CeH. Male carriers met the biochemical criteria of CeH, and FT4 concentrations of all female carriers were in the lower half of the reference interval (figure 3). Six of seven male carriers had blunted TRH test results. All carriers had relatively low thyroid volumes, in keeping with relative TSH deficiency. While most carriers had T3 concentrations within the age-appropriate reference intervals, adult T3/T4 ratios were slightly higher compared with euthyroid subjects.18 Although T3/T4 ratios have not been reported in untreated patients with CeH, untreated TBL1X mutation carriers also had T3 concentrations within reference interval combined with low-normal T4 concentrations, suggesting a slightly increased ratio.5 These findings may help distinguish CeH from the non-thyroidal illness syndrome, which is associated with reduced serum T3 concentrations,23 and thus decreased T3/T4 ratios. All male carriers had cholesterol and SHBG levels within the reference interval, although this was measured during treatment with LT4, while three female mutation carriers had SHBG concentrations below the reference interval. Two of these women had elevated total cholesterol levels. Since low levels of SHBG and elevated lipid levels are associated with hypothyroidism,24 25 this may point to a certain degree of hypothyroidism at the target tissue level, although the number of observations is too small, and the variation in the population is too large to draw any firm conclusions at this stage.
Mutations in IRS4 have not been associated with CeH before. A missense mutation has been described in a female with paranoid schizophrenia, but thyroid function was not reported in this patient.26 In addition, IRS4 mutations have been associated with a higher BMI in patients with schizophrenia,27 and IRS4 was found to be associated with body size in dogs.28 Only three loss-of-function mutations in IRS4 are reported in the ExAC database, none of which were homozygous or hemizygous. Although two other genes associated with isolated CeH, TBL1X and IGSF1, are also located on the X-chromosome, they are distant from IRS4, with IRS4 located on Xq22.3, TBL1X on Xp22.3-p22.2 and IGSF1 on Xq26.1.
IRS4 is located on the X-chromosome and encodes a 1257 amino acid protein, which contains a pleckstrin homology (PH) domain and a phosphotyrosine-binding (PTB) domain in the N-terminal region, followed by multiple tyrosine and serine phosphorylation sites in the C-terminal tail.29 IRS4 is part of the IRS family, which contains six members (IRS1–6). All proteins contain PTB domains and multiple glycosylation sites to induce a cascade in hormone signalling pathways.29 The best characterised are IRS1 and IRS2, which are involved in insulin signalling and of which knockdown results in marked dysregulation of growth and metabolic homeostasis.30 31 The function of IRS3-6 is still uncertain but thought to be highly specific due to their restricted tissue distribution.32 IRS3 and IRS4 were found to influence actions of IRS1 and IRS2.33 Although IRS1 and IRS2 were found to be non-redundant, function interchangeability was found between IRS1 and IRS3. The functional overlap between other family members is still under investigation.32
The IRS4 protein binds to and is activated by the intracellular domain of tyrosine kinase receptors, most notably the insulin, IGF-1 and leptin receptors (IR, IGF1R and LEPR).6 IRS4 mRNA is expressed in the pituitary gland and hypothalamus,7 skeletal muscle, heart, kidney and liver of rats.34 In human tissue, RNA expression was detected in hypothalamus, pituitary, thyroid and ovary tissue (http://www.proteinatlas.org/ENSG00000133124-IRS4/tissue; accessed 6 May 2017). In the present study, we confirmed IRS4 mRNA expression in the human pituitary gland and found expression in a number of hypothalamic nuclei, including the paraventricular nucleus. Although we did not measure IRS4 protein expression, marked RNA expression suggests the presence of protein in those tissues.
IRS4 KO mice were reported to exhibit mild metabolic differences compared with WT mice. Particularly males were slightly smaller, exhibited lower blood glucose levels in fasted and fed states and had an impaired OGTT. Additionally, KO pairs reproduced less well, attributed to decreased fertility of females. Their thyroid function was not reported.35
In this study, we found that Irs4 KO mice did not have reduced serum TSH or thyroid hormone concentrations, despite clearly decreased Tshb mRNA expression in the pituitaries of KO females. In addition, we found no significant effect of IRS4 absence on Trh expression in the hypothalamus. The finding that Tshb mRNA expression was decreased only in female Irs4 KO mice and that female KO and WT mice had somewhat lower TSH concentrations than male KO and WT mice may suggest the presence of sexual dimorphism in the HPT axis. Although other characteristics of Irs4 deficiency seem more pronounced in males,35 this may suggest that female mice require Irs4 for sufficient Tshb transcription, while males do not. The mouse data seem to contradict the human findings, suggesting that KO mice may not be an optimal model. IRS4 expression may vary between species and have different contributions to the regulation of the HPT-axis. A similar situation has been reported for other genes; one example is the mouse model for IGSF1 deficiency, which does not show the decreased serum total thyroid hormone concentrations seen in human patients.36 This also holds true for MCT8, a TH transporter; patients with a mutation in MCT8 have a serious neurologic phenotype (Allan–Herndon–Dudley syndrome), while MCT8 KO mice behave normally.37
Although impaired glucose tolerance was reported in Irs4-null mice,35 in this study, IRS4 mutation carriers did not show signs of glucose intolerance. All patients with IRS4 mutations had a normal height and weight. One female carrier underwent fertility treatment for fallopian tube obstruction. Due to the association of IRS4 with IR, IGF1R and LEPR, we considered the possibility that mutations in IRS4 impair the signalling capability of these receptors. However, patients in this study had no abnormalities in either OGTT or HOMA, suggesting a normal insulin sensitivity, nor did they exhibit growth failure. Furthermore, thyroid function in patients with INSR or IGF1R mutations has been reported as being normal.38 39 These findings make a role for IR and IGF1R in the pathogenesis of CeH in patients with IRS4 mutations unlikely.
The combination of these findings emphasises the complexity of the central regulation the HPT axis and indicates that further research is necessary before a (putative) mechanism explaining the decreased plasma T4 concentrations in patients harbouring a mutation in IRS4 can be proposed. For instance, testing of the HPT axis in a more dynamic way in mice and other species with mutations more similar to those in the human patients may offer more insight.
Hypothalamic leptin signalling is closely associated with thyroid function. During starvation, decreasing leptin concentrations lead to reduced TRH expression in the paraventricular nucleus, contributing to persistently low plasma TSH and thyroid hormone concentrations. This response to fasting may function to conserve energy.40 Leptin acts on the HPT axis directly through receptors on hypophysiotropic TRH neurons that upregulate proTRH production and conversion to TRH40 and indirectly through pro-opiomelanocortin and neuropeptide Y/agouti-related peptide expressing neurons in the hypothalamic arcuate nucleus that project to TRH neurons.41 In healthy men who were fasted for 72 hours, 24-hour TSH secretion studies showed normal baseline concentrations but marked suppression of TSH secretion and loss of typical pulsatility characteristics. The fasting-induced decrease in TSH pulsatility was restored by leptin administration.42 Additionally, 56-hour fasting in healthy men and women was shown to decrease TSH responses to TRH by 43% below baseline responses.43 In the present study, IRS4-mutated patients showed remarkably similar TSH baseline concentrations, 24-hour TSH secretion patterns and TRH test results. These data may suggest that the central downregulation of the HPT axis in patients with IRS4 mutations is mediated by subtle hypothalamic LEPR dysfunction. Leptin-deficient patients have a highly heterogeneous phenotype, with case-series describing subclinical hypothyroidism with increased TSH and normal FT4 levels,44 normal thyroid function45 and normal TSH concentrations combined with relatively low FT4 concentrations, which increased after treatment with r-metHuLeptin.46 TRH tests in these patients were normal and exaggerated in a subclinical hypothyroid patient.44 Interestingly, 24-hour TSH secretion studies in leptin-deficient patients found suppression of TSH secretion46 and variable loss of organisation of TSH pulsatility which, in one study, was strongly correlated to the 24-hour leptin secretion.47 Leptin-resistant patients were described to have either normal thyroid function, or low levels of FT4 combined with normal levels of TSH, similar to patients with mutations in IRS4. However, these patients showed an exaggerated and sustained TSH response to TRH, while IRS4 mutation patients had a blunted response to TRH.48 Whether IRS4-mutated patients may have LEPR dysfunction is highly speculative at this time, and the mechanism behind the development of CeH in these individuals requires further investigation.
An interesting topic for further investigation is TSH bioactivity in patients with IRS4 mutations. Decreased TSH bioactivity has been described in several Mendelian causes of isolated CeH, such as mutations in TSHB and TRHR.1 Whether this is also the case in patients with IRS4 mutations remains to be studied.
In conclusion, we identified IRS4 mutations in patients with congenital isolated CeH. Although Irs4 KO mice did not display CeH, the human data suggest that IRS4 represents a novel genetic cause of isolated CeH.
Acknowledgments
The authors are grateful for the critical review and advice of Professor D J Bernard, McGill University, Montreal; the excellent help and advice of E Endert, Laboratory of Endocrinology, AMC, Amsterdam; and for the technical assistance of O V Surovtseva, Laboratory of Experimental Endocrinology, AMC, Amsterdam. We would like to thank the patients and their families who kindly consented to participate.
References
Footnotes
Senior authors AB, ASPvT and EF contributed equally.
Contributors CAH coordinated the study. CAH, RCH, AB, ASPvT and EF designed the study. EMdV, NZ-S, ELTvdA, BB, GH-N and ASPvT provided patient care and collected data. MA and HB performed genetic analysis. FR performed statistical analysis. CAH, RCH, AB, ASPvT and EF interpreted the results and wrote the manuscript. All authors critically reviewed the report. No writing assistance was provided. CAH, AB, ASPvT and EF had full access to all of the data in the study and take responsibility for the integrity of the data. All authors revised the manuscript critically and approved the final version for publication.
Funding This work was financially supported by an AMC Foundation grant.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The Medical Ethics Committee of the Academic Medical Center, Amsterdam, approved the study protocol (NL52353.018.15).
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it was published Online First. A statement about equal contribution has been added to the first page.