Article Text

Statistics from Altmetric.com
- BN-PAGE, blue native polyacrylamide gel electrophoresis
- CS, citrate synthase
- CT, computed tomography
- LHON, Leber’s hereditary optic neuropathy
- MELAS, mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes
- MRI, magnetic resonance imaging
- mtDNA, mitochondrial DNA
- mt-tRNA, mitochondrial transfer RNA
- RFLP, restriction fragment length polymorphism
Complex I is the largest of the mitochondrial respiratory chain enzyme complexes, consisting of at least 46 subunits, seven of which are encoded by mtDNA. Deficiency of complex I is the most common respiratory chain defect, and can be caused by mutations in both nuclear and mtDNA encoded genes. It has a wide range of clinical presentations, from lethal infantile mitochondrial disease to isolated myopathy.1–3 Mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) is one of the syndromes associated with complex I deficiency and in approximately 80% of cases is caused by a mutation, 3243A→G, in the mitochondrial tRNALeu(UUR) gene (MTTL1). Other mutations in MTTL1 and other transfer RNA genes (MTTF, MTTV, MTTQ) account for most of the remainder of cases.4 However, a number of mutations in the mitochondrial MTND subunit genes of complex I have also been reported to cause MELAS, most notably in MTND55 and to a lesser extent in MTND6.6
In stark contrast, there are presently no mutations in the MTND1 subunit gene associated with MELAS. There are several mutations in MTND1 associated with Leber’s hereditary optic neuropathy (LHON) which may be pathogenic, but only one, the 3460G→A mutation, that has robust evidence, including cell biology studies, for pathogenicity.7–9 Here we report three unrelated patients with MELAS and isolated complex I deficiency in skeletal muscle and cultured fibroblasts due to previously unreported mutations in the MTND1 gene. Evidence confirming the pathogenic nature of these mutations includes data from cell fusion experiments and blue native polyacrylamide gel electrophoresis (BN-PAGE), the latter confirming a crucial role for the ND1 subunit in the assembly of complex I holoenzyme.10
PATIENTS
Patient 1
Patient 1, a white male, presented at 4 years of age with a 3 month history of increasing tiredness, clumsiness, frequent falls, and weakness of his left side. On examination he had increased tone and brisk reflexes in his left leg and walked with a hemiplegic gait. Magnetic resonance imaging (MRI) revealed lesions of the right lentiform and caudate nuclei. He made a partial recovery, but at 7 years of age presented with right sided facial twitching and speech difficulties. EEG suggested focal seizures, and MRI revealed new lesions of the right thalamus, left basal ganglia, and bifrontal cortical regions. Magnetic resonance spectroscopy revealed increased levels of lactate in these areas. CSF and blood lactate levels were 2.7 mmol/l and 1.3 mmol/l respectively (normal <2.5 mmol/l for both). Skeletal muscle histology and enzyme histochemistry were normal, but electron microscopy indicated focal subsarcolemmal accumulation of mitochondria. The seizures resolved over 6 months. At 9 years of age he had a period of lethargy, slurred speech, and relatively poor functioning lasting 2 months. MRI revealed extension of the lesions in the left basal ganglia and right frontal cortex. He has had no further acute episodes and at 10 years old he walks independently but requires a wheelchair for long distances. He has retained normal intelligence and there is no family history of note. On examination he has no cranial nerve signs and normal proximal power, but increased tone and brisk reflexes, especially in the lower limbs.
Key points
-
Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) is one of many clinical presentations associated with mutations in mitochondrial DNA (mtDNA) and a biochemical deficiency of respiratory chain complex I. While the ‘common’ 3243A→G mutation in the mitochondrial tRNALeu(UUR) (MTTL1) gene is detected in approximately 80% of patients with MELAS, the aetiology of the remaining 20% is heterogeneous, with different causative mutations occurring in MTTL1, other mitochondrial tRNA (mt-tRNA) genes, and the MTND genes encoding subunits of complex I.
-
We report three unrelated patients with MELAS in whom the 3243A→G mutation could not be detected, but who expressed a specific deficiency of complex I activity in both skeletal muscle and cultured fibroblasts. Targeted sequencing of mt-tRNA and MTND genes revealed that each patient harboured a different novel mutation in the MTND1 subunit gene of complex I (3697G→A, 3946G→A, and 3949T→C), each of which predicted an amino acid change. The failure to restore complex I activity upon fusion of patient cells with a cell line lacking mtDNA (ρ0 cells) confirmed pathogenicity for each of these mutations, all of which appear to perturb the assembly or turnover of complex I.
-
Our finding of three novel mutations in the MTND1 gene causing MELAS is supportive of recent papers highlighting the importance of mtDNA mutations in the aetiology of childhood respiratory chain complex I deficiency and underlines the importance of a cohesive diagnostic strategy where molecular genetic investigations are shaped by the biochemical and histological findings.
Patient 2
Patient 2, a white female, had normal developmental milestones until 5 years of age, when anxiety issues and schooling difficulties became evident. From 11 years of age she developed hearing problems, and high tone deafness was confirmed at when she was 14 years old. She presented at 14 years of age with frontal headaches, vomiting, and multiple left sided focal seizures, which became generalised over a 24 hour period. MRI and computed tomography (CT) of the head showed a right parietal lobe ‘watershed’ infarct, which showed some progression 7 days after the onset of seizures. Subsequent MRI and CT scans revealed further ischaemic areas in both occipital regions, and in the parietal, temporal, and frontal lobes. CSF and plasma lactate levels were elevated at 10.3 mmol/l and at 3.5 mmol/l respectively. Skeletal muscle biopsy was normal on both light and electron microscopy. Recurrent strokes complicated by seizures, episodes of cortical blindness of variable completeness, and other neurological deficits have occurred, leaving her with a moderate intellectual disability. A maternal half sister and full sister are reported to tire easily, as does her mother, who also suffers from high tone deafness and mild glucose intolerance.
Patient 3
This male patient of Chinese origin presented at 14 years of age with a short history of left sided upper limb focal seizures. The EEG showed focal spike and slow wave discharges over the right parasagittal region. A few months later, he presented with headache and blurred vision. On examination he had a left homonymous hemianopia with macular sparing, and MRI showed lesions in the right occipital and posterior frontal cortex. His visual problems resolved, but over the following year he had episodes of altered sensation in his limbs. A further MRI showed new T2 hyperintensity lesions in the anterior aspect of the left occipital lobe, the medial right parietal cortex, and the anterior frontal cortex. The basal ganglia were not affected. His CSF and plasma lactate levels were 3.4 mmol/l and 3.0 mmol/l respectively. Skeletal muscle enzyme histochemistry showed subsarcolemmal accumulations of mitochondria and EM revealed a moderately increased number of large, abnormally shaped mitochondria with abnormal cristae and excess lipid. At 16 years of age he has normal intelligence and little of note on examination. He has regular episodes of focal left hand seizures that are difficult to control but have not generalised. Medications include carbamazepine, coenzyme Q10, riboflavin, and vitamin C. His mother and an older sister suffer from migraine, otherwise there is no family history of note.
METHODS
Biochemistry
The activities of the individual respiratory chain complexes and the mitochondrial matrix marker enzyme citrate synthase were measured in skeletal muscle homogenates and mitochondrial fractions isolated from cultured cells as described previously.2,11
mtDNA analysis
Total DNA was extracted from tissues, cultured cells, urine sediment, and blood by standard procedures. Manual sequencing of the MTTL1 gene was performed using a ThermoSequenase radiolabeled terminator cycle sequencing kit (USB Corporation, Cleveland, OH, USA). For automated sequencing, the coding region of the mitochondrial genome was amplified using a series of overlapping oligonucleotide primer pairs and sequenced with a BigDye terminator cycle sequencing kit as described previously.12 Sequence data were analysed using Navigator and Factura software (Applied Biosystems, Warrington, UK) and compared to the revised Cambridge reference sequence of human mtDNA.13
Quantification of mtDNA heteroplasmy
Putative pathogenic sequence variants in tissues and cells from the patients and other family members were further investigated by last hot cycle PCR-restriction fragment length polymorphism (RFLP) analysis. Details of the oligonucleotide primers and restriction endonucleases used for each of the patients are provided in table 1, along with the expected fragment sizes after digestion. The relative proportions of normal and mutated mtDNA genomes in different tissues were determined by the addition of 5μCi [α-32P]-dCTP (3000 Ci/mmol) prior to the last cycle of the PCR. Labelled products were digested with 10 U of the appropriate restriction endonuclease (table 1), separated through a 12% non-denaturing polyacrylamide gel, and the radioactivity in each fragment quantified using ImageQuant software (Molecular Dynamics, Sunnyvale, CA, USA).
PCR-RFLP analysis of putative pathogenic MTND1 gene mutations in the three patients
Cell biology studies
Cells were cultured in Dulbecco’s modified Eagle’s medium (JRH Biosciences, Lenexa, KS, USA), supplemented with 10% fetal calf serum (JRH Biosciences) and 50 μmol/l uridine.
Generation of antibiotic resistant cell lines
G418 resistance and increased growth potential were conferred on a control fibroblast cell line and the 143BTK-ρ0 osteosarcoma cell line as described.14 Similarly, patient fibroblasts were transfected with Hygromycin B resistance and SV40 T antigen via pGKHygT, derived from the pGKHyg plasmid (gift of Dr R Saffery). Antibiotic resistant clones were isolated after selection with Hygromycin B (Roche Diagnostics Australia, Castle Hill, NSW, Australia).
Generation of hybrids
Two cell lines with different antibiotic resistance were co-cultured until confluent and fused using polyethylene glycol (PEG 1500, 50% solution in 15 mmol/l HEPES; Roche Diagnostics Australia, Castle Hill, NSW, Australia). Heterokaryons were selected in medium containing both G418 (200 μg/ml) and hygromycin B (100 μg/ml). Dual antibiotic resistant hybrids were passaged in the selective medium at least twice after the parental and “self self” fused cells had died (usually after 5–10 days) before analysis of complex I activity. Cell fusion experiments and complex I enzyme analysis were performed in duplicate.
Protein analysis
The amount and size of assembled complex I was studied by BN-PAGE and immunoblotting using mitochondria isolated from primary fibroblast cell lines from patients 1, 2, and 3 as described previously.14,15
RESULTS
Biochemistry
Respiratory chain enzymes in skeletal muscle homogenates and cultured skin fibroblasts from each patient were consistent with an isolated defect of complex I according to the criteria described previously2 (table 2). For patient 1, complex I activity in muscle was not clearly deficient when expressed relative to protein or as a ratio to the mitochondrial matrix enzyme citrate synthase (CS), but was deficient when expressed relative to complex II (23% of control mean). All respiratory chain enzymes measured except complex I were 180–280% of control means relative to CS in that sample, reflecting mitochondrial proliferation.
Residual complex I activities in skeletal muscle homogenates and isolated fibroblast mitochondria
Identification of pathogenic mutations
Sequence analysis of the MTTL1 gene was normal for all three patients. This prompted sequencing of the ND and mt-tRNA coding regions of the mitochondrial genome. In addition to recognised polymorphisms, each patient had a number of changes that had not been reported previously in public databases16,17 (see also http://www.genpat.uu.se/mtDB/polysites.html). While many changes were synonymous or occurred in non-conserved regions of the genome, each patient had a different heteroplasmic mutation, predicted to change a conserved amino acid of the MTND1 gene. These mutations, (3697G→A (G131S), 3946G→A (E214K) and 3949T→C (Y215H)), were therefore potentially pathogenic and entirely consistent with a resultant complex I deficiency.
PCR-RFLP assays were developed for each of these sequence variants, and mutant loads were determined by last cycle hot PCR (table 1). Patient 1 had 80% and 79% of the 3697G→A mutation in skeletal muscle and fibroblasts respectively. The mutant load was 3% in blood from his mother, but no mutation was detected in blood from his two sisters (fig 1A), two maternal aunts, maternal uncle, or maternal grandmother (not shown), all of whom were asymptomatic.

Quantification of the level of mutated mtDNA in patients and other relatives by PCR-RFLP analysis. Details of the oligonucleotide primers, restriction endonucleases and expected digestion products are given in table 1. (A) Patient 1 (3697G→A); (B) patient 2 (3946G→A); (C) patient 3, (3949T→C). U, uncut sample; C, control; M, skeletal muscle; F, fibroblasts; B, blood. Lanes 1–3 show blood from the mother and two sisters of patient 1; lanes 4–6 show blood and urinary sediment from the mother and urinary sediment from the sister of patient 2; lanes 7–10 show blood taken from the mother, father, and two brothers of patient 3.
Patient 2 harboured somewhat lower mutant load, with 60% of the 3946G→A mutation in muscle, 45% in fibroblasts, and 37% in blood. No mutation was detected in blood or urine sediment from her mother, while 3% mutant load was detected in urine sediment from her sister (fig 1B), implying that although the mother harbours the mutation in ovarian tissue, it may be selected against in mitotic tissues as previously reported for the 3243A→G MTTL1 mutation.18
Patient 3 had 93% of the 3949T→C mutation in muscle, 88% in fibroblasts, and 45% in blood. Thus it appears likely that the 3949T→C mutation is also selected against in blood. No mutation was detected in blood from his asymptomatic mother and two brothers (fig 1C).
We also used PCR-RFLP analysis to screen for the 3697G→A, 3946G→A, and 3949T→C mutations in 68 complex I deficient paediatric patients in whom no causative mutation had been found. None of these mutations was detected in these patients.
Cell biology studies
When fibroblasts from a complex I deficient patient with a presumed nuclear DNA defect were fused with the 143BTK-ρ0 osteosarcoma cell line, the hybrids showed restoration of complex I activity. However, fusion of fibroblasts from the three patients reported here and the 143BTK-ρ0 osteosarcoma cell line failed to restore complex I activity (fig 2). As the ρ0 cell line contains no mtDNA, lack of phenotypic rescue in the hybrids indicates that the nucleus of the ρ0 cell line is unable to correct the complex I defect. These results imply that the complex I defect in the three patient cell lines is of mtDNA origin19,20 and confirm that the novel mutations described here are causative.
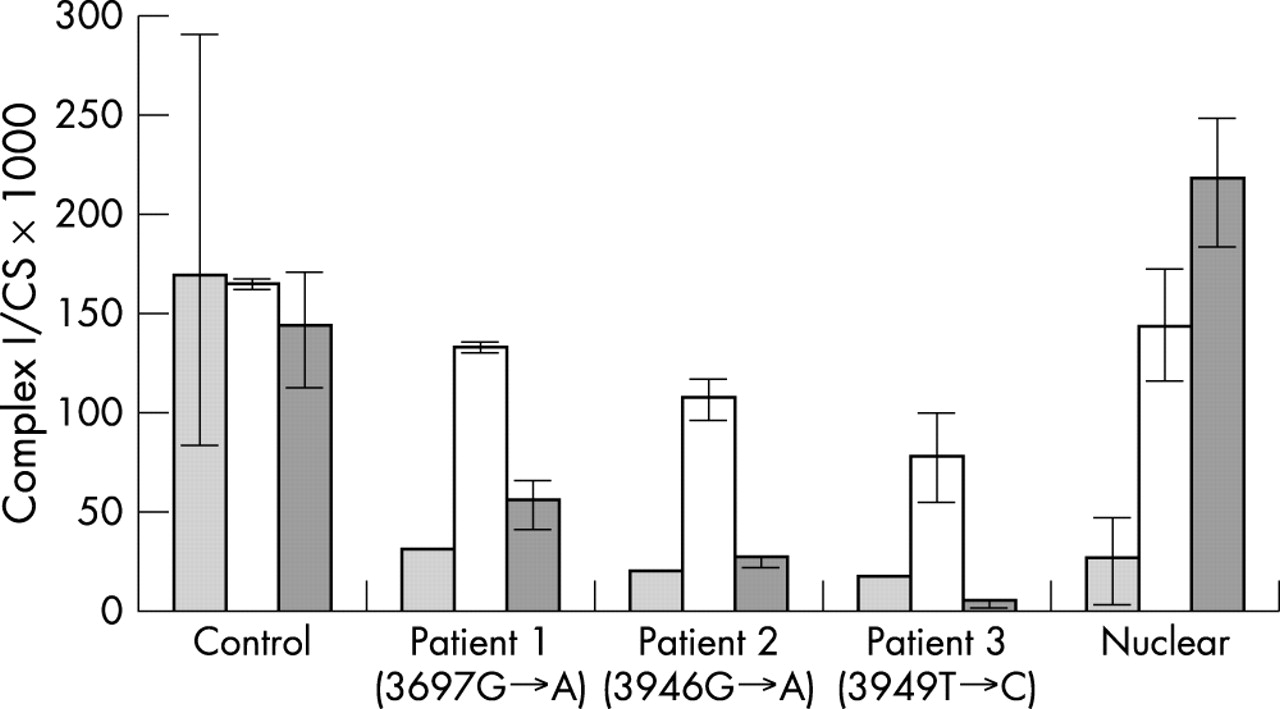
Complex I activity (expressed relative to citrate synthase; CS) in patient cell lines and hybrids. The light grey columns show complex I activity in transformed fibroblasts. Open bars show complex I activity in hybrids between control transformed fibroblasts and transformed fibroblasts from either a control, patient 1, 2, 3, or a patient from a consanguineous pedigree with a presumed nuclear DNA mutation. Dark grey columns show complex I activity in hybrids between the 143BTK-ρ0 osteosarcoma cell line and the control or patient cell lines. The error bars represent the observed range for all complex I measurements for that cell line, which were as follows: control transformed fibroblast cell line (n = 23); parental transformed fibroblasts from patients 1, 2, and 3 (n = 1); “nuclear” patient transformed fibroblasts (n = 8); patient 1×ρ0 hybrid (n = 4); and all other fusions (both with control and ρ0 cell lines) (n = 2).
Protein studies
Analysis of the intact complex after separation by BN-PAGE with an antibody to the 39 kDa subunit of complex I revealed dramatically reduced levels of fully assembled complex I for each of the three patients (25% for 3697G→A, 11% for 3946G→A, and 12% for 3949T→C) compared to a control fibroblast cell line (fig 3).

Analysis of levels of assembled complex I by BN-PAGE. Mitochondria isolated from patient fibroblasts and a control (Con) were solubilised in dodecyl maltoside and subjected to BN-PAGE and subsequent Western blot analysis using antibodies specific for the complex I 39 kDa subunit and complex II 70 kDa subunit. The amount of fully assembled complex I relative to complex II was estimated by densitometry of the BN-PAGE immunoblot and shown to be 25% for patient 1, 11% for patient 2, and 12% for patient 3.
DISCUSSION
All three patients presented with a classical MELAS phenotype and suggestive family history, but without evidence of 3243A→G or other mutations in the MTTL1 gene. The finding of an isolated complex I deficiency, with normal complex IV/CS and complex IV/complex II ratios in skeletal muscle, led us to investigate a possible mtDNA mutation as the cause of the clinical disease. The elevation of complex II observed in muscle from patients 1 and 3 is entirely consistent with the mitochondrial proliferation noted in their muscle biopsies, a frequent histological finding in many mtDNA disorders.
Sequencing revealed a different novel mutation in the MTND1 subunit gene of each patient. These mutations were heteroplasmic in skeletal muscle and cultured skin fibroblasts, where the mutant load also correlated with the complex I defect. Patient 2 had a somewhat lower mutant load (60% in muscle and 45% in fibroblasts), but this is not without precedent in complex I genes, as comparably low mutant loads of the 13513G→A (MTND5) have been associated with Leigh disease.14 The mutations were not present in three large mtDNA sequence databases (see Results) nor were they present in 68 complex I deficient children in whom no causative mutations had been identified. Pathogenicity was confirmed by the failure to restore complex I activity upon fusion of fibroblasts with a cell line (ρ0) containing no mtDNA (fig 2). In these circumstances, patient mitochondria are observed in a different nuclear background and rescue of the biochemical defect is therefore evidence of a nuclear origin for the complex I deficiency.19,20
At present, there is only one mutation in the MTND1 gene with compelling evidence for pathogenicity, 3460G→A, which is thought to cause LHON.7–9 We believe the novel mutations described here represent three further pathogenic mutations in MTND1. Firstly, they all change conserved amino acids of complex I and are associated with complex I deficiency in patient tissues and in ρ0 hybrids. Secondly, they are heteroplasmic, with much higher levels of the mutation present in patient tissues than in unaffected relatives. Finally, they are absent both in large numbers of controls and in asymptomatic maternal relatives of the probands.
ND1 is one of the most conserved complex I subunits.21 Situated in the hydrophobic arm of the L shaped complex I protein, it forms eight transmembrane spanning domains that are postulated to be involved in ubiquinone binding and proton pumping,22 processes known to be disrupted by mutations in homologous bacterial genes. Specific impairment of ubiquinone reductase activity has been demonstrated following mutation of the nqo8 gene (homologous with human ND1) of Paracoccus denitrificans.23 Furthermore, studies of the NQO8 subunit suggest that glutamate residues located close to helices E and F on the matrix side of the mitochondria are involved in quinone binding and reduction.24 In Rhodobacter capsulatus, the ND1 homologue NUOH and the NUOD subunit, which is the homologue of the nuclear encoded 49 kD subunit of complex I in mitochondria, also appear to be involved in ubiquinone binding.25 Interestingly, the three novel mutations reported here, in common with mutations described in bacterial homologues of MTND1 and the LHON mutation 3460G→A, also cause an amino acid change in the hydrophilic loops that face the mitochondrial matrix (fig 4). The 3697G→A (G131S) mutation in patient 1 occurs at a highly conserved residue in the loop between transmembrane domains C and D.21,26 The 3946G→A (E214K) and 3949T→C (Y215H) mutations in patients 2 and 3 are in the glutamic acid rich hydrophilic loop between transmembrane domains E and F, one of the most conserved regions of the ND1 subunit.21,26 The mutations in patients 2 and 3 change the charge of the amino acid and are both predicted to abolish a β turn in the ND1 protein.27

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Predicted structure of human ND1 showing the eight transmembrane spanning domains (http://sosui.proteome.bio.tuat.ac.jp). The three novel mutations in patients 1, 2, and 3 (shown in bold), together with the pathogenic 3460G→A LHON mutation are indicated, highlighting their positions within conserved, hydrophilic, extramembrane loops. The corresponding amino acid sequences from several species surrounding the mutations (from residues 125–140 and 192–229 of the human sequence) are shown. The three residues affected by the novel mutations (in bold) are highly conserved.
At the protein level, the mutations in our MELAS patients appear to affect complex I assembly or turnover (fig 3). The BN-PAGE results demonstrate low levels of fully assembled complex I in fibroblasts from the three patients, with the 3946G→A (E214K) and 3949T→C (Y215H) mutations in patients 2 and 3 appearing to be more detrimental. The ND1 subunit is thought to be incorporated into the complex I structure at an early stage of its assembly.10 This implies that the decreased levels of fully assembled complex I are due to impaired assembly of complex I, with consequent degradation of unincorporated subunits. A similar mechanism has been demonstrated for the cytochrome c oxidase subunits in patients with mutations in SURF1.28
It is interesting to note that all three patients with MTND1 mutations expressed a complex I defect in cultured fibroblasts. Similarly, we found that all of eight patients with mutations in the MTND3,15MTND5,14 and MTND629 subunit genes who presented with Leigh disease or other infantile encephalopathies also expressed complex I deficiency in fibroblasts. By contrast, our experience with five patients with complex I deficiency caused by mutations in MTTL1 (two with 3243A→G and one each with 3250T→C, 3303C→T, and 3242G>A)2 is that all but the 3242G>A patient had low mutant loads and normal complex I activity in fibroblasts (results not shown). Thus patients with mutations in MTND subunit genes express a complex I defect in fibroblasts more often than those with MTTL1 mutations.
Liolitsa et al have proposed that the MTND5 gene is a hot spot for mutations causing MELAS, and note four mutations in this gene that have been associated with the syndrome: 13513G→A, 13514A→G, 12770G→A, and 13045A→C.5 Certainly, it would appear that the aetiology of MELAS is diverse and that following exclusion of the 3243A→G mutation, a targeted sequencing approach should be directed by the results of muscle biochemistry. In patients with MELAS, isolated complex I deficiency has proved an important indicator of a MTND gene mutation and our finding of three novel mutations in three unrelated patients suggests that the MTND1 gene may also be a hot spot for mutations that cause this devastating disease.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council of Australia, the Australian Research Council, the Muscular Dystrophy Association of the USA, the Wellcome Trust, the Muscular Dystrophy Campaign UK and the Newcastle upon Tyne Hospitals NHS Trust. R McFarland is an MRC Clinician Scientist and D R Thorburn is an NHMRC Senior Research Fellow.
REFERENCES
Footnotes
-
Conflicts of interest: none declared