Article Text

Statistics from Altmetric.com
Editor—The most common sensory deficit in humans is hearing loss, affecting 1 in 1000 children, with approximately half of the cases having a genetic cause. The majority of these genetic causes are non-syndromic, of which approximately 75% have an autosomal recessive mode of inheritance.1 So far, nearly 30 genes that cause non-syndromic recessive deafness (NSRD) have been located (for review see http://dnalab-www.uia.ac.be/dnalab/hhh). The loci corresponding to NSRD are designated DFNB, with a number corresponding to the chronology of their localisation. The first locus, DFNB1 (MIM 220290), located on chromosome 13q11-12,2 has been shown to be responsible for nearly half of NSRD owing to mutations in the gene encoding the gap junction protein connexin 26 (GJB2) (MIM 121011).3 4 One mutation, 30delG (also referred to as 35delG), accounts for the majority of mutations in this gene in some ethnic groups,3-8 while it is rarer in others.9-15
The geographical position of Lebanon, a small country of 10 500 km2 on the eastern shores of the Mediterranean sea, has made it a historical crossroads between Asia, Africa, and Europe. As a consequence, the Lebanese population shows a wide genetic diversity, with no less than 17 ethno-religious communities. Today, the population is approximately 4 million people, with a world wide diaspora estimated at 15 million. In Lebanon, consanguineous marriages are still frequent (from 10 to 30%), favouring the incidence of autosomal recessive diseases,16 such as haemoglobinopathies, sickle cell anaemia, familial Mediterranean fever, congenital hypothyroidism, cystic fibrosis, and deafness.
The purpose of this study is to summarise the different NSRD loci found in a case series of Lebanese families and to determine the carrier frequency of the 30delG mutation in selected Lebanese subjects.
Material and methods
Forty eight multiplex Lebanese families with non-syndromic congenital moderate to profound deafness, from various regions of the country, were included in this analysis. Informed consent was obtained from each family member before clinical investigation, blood sampling, and DNA analyses. Hearing loss was documented by audiological testing and a thorough examination was conducted to ensure the absence of any clinical sign suggesting another diagnosis. The pattern of inheritance was autosomal recessive for all the families studied.
The methods used for DNA extraction and linkage analyses were the same as those presented elsewhere.17-23 In families linked to the DFNB1 locus, affected subjects were first tested for the common 30delG mutation according to Storm et al.8 In the absence of this mutation, amplification of the coding exon of the GJB2 gene and DNA sequencing was performed according to Denoyelle et al.3
Additionally, 300 unrelated Lebanese subjects with no familial history of hearing problems, selected from our laboratory database to represent all ethno-religious Lebanese communities, were analysed to assess the prevalence of the 30delG mutation. Informed consent was obtained from each person involved and DNA was extracted from peripheral blood leucocytes following standard methods.24
Results
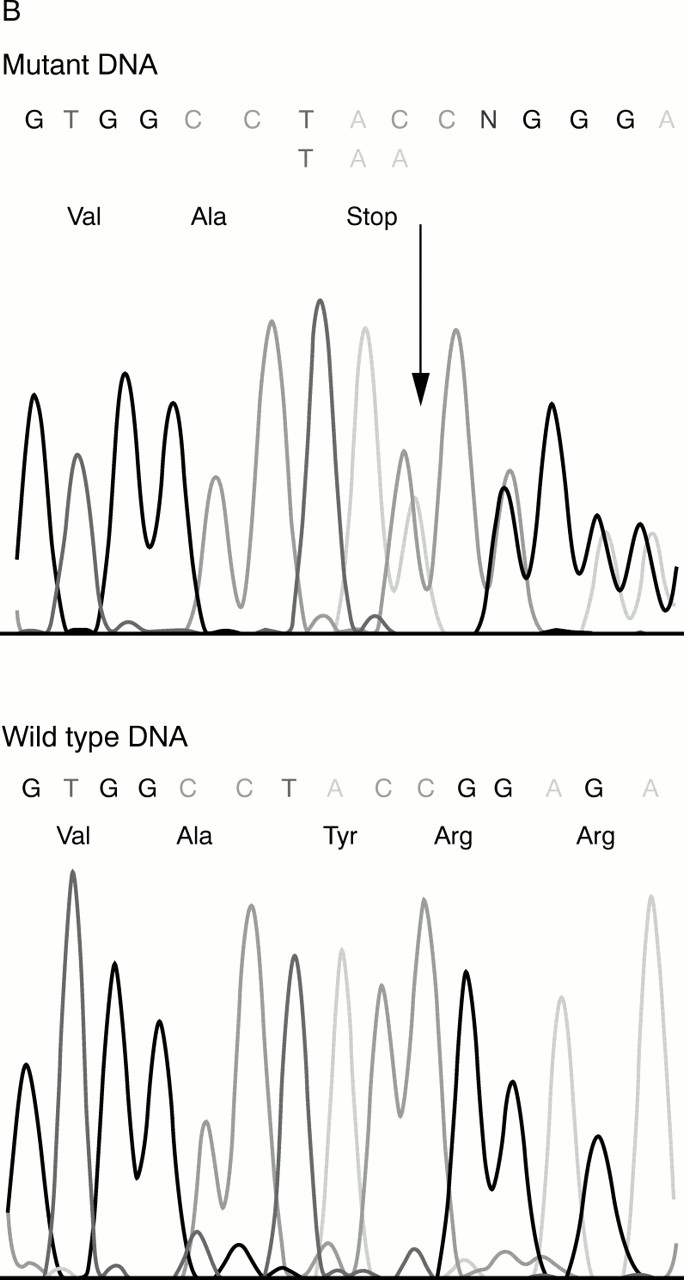
Of the 48 Lebanese pedigrees investigated with non-syndromic recessive moderate to profound deafness, 37 were Muslims, 10 Christians, and one Druze. Nearly 92% (44) of them were consanguineous. Table 1 summarises the different loci found in our population. Sixteen of these families were linked to the DFNB1 locus (33%). In all of them, the affected subjects had a severe to profound hearing loss. In 15 of them, affected subjects were homozygous for the 30delG mutation in the GJB2 gene. In the last one, a non-consanguineous Christian Maronite family, affected subjects were compound heterozygotes for two novel mutations, a missense mutation, replacing arginine by histidine at codon 32 (R32H), and an insertion of an adenine in position 291 (291insA) creating a stop codon (fig 1A, B).
Different loci found in 48 Lebanese families with autosomal recessive non-syndromic hearing loss

{kind=link}
{kind=link}
DNA sequencing of the GJB2 coding exon showing (A) a missense mutation, replacing arginine by histidine at codon 32 (R32H), and (B) an insertion of an adenine in position 291 (291insA) creating a stop codon.
In the remaining 32 families, at least 13 implicated loci were found (table 1). In three of these families, it was not possible to determine exactly which of three loci, DFNB4, 14, or 17, were implicated. They are all localised on the long arm of chromosome 7 at q31, and these three families were not sufficiently informative to allow a precise localisation. In four families, linkage to DFNB9, 10, and 12 was highly likely but could not be proven since the lod score was below 3 at θ=0.00. Finally, seven families have not been linked to any of the already reported loci so far and are still undergoing more elaborate analysis.
In the 300 subjects tested for the 30delG mutation, we found seven heterozygotes, four Christians and three Muslims. Four were men and three women. This mutation was found with an allele frequency of 1/86, giving a prevalence of 1/7400 newborns affected with deafness secondary to a 30delG mutation.
Discussion
The majority of the cases of hereditary deafness are non-syndromic. So far, at least 77 chromosomal loci have been identified, 39 for autosomal dominant DFNA, 30 for autosomal recessive DFNB, and eight for X linked DFN1(http://dnalab-www.uia.ac.be/dnalab/hhh). The contribution of theGJB2 gene in NSRD in Lebanon (33%) is lower than in other studies around the world: 49% in Italy and Spain,6 40% in the USA,25 and 41.3% in Israel.26 However, the allele frequency of the 30delG mutation among our familial DFNB1 cases was ∼94%. Although, our sample size is relatively small, we suggest that this allele frequency is higher than in European populations where it has been estimated at 60-80% of all known GJB2mutations.14 The haplotype of affected subjects from most of these families was different (data not shown) suggesting that this mutation arose independently in each family. Furthermore, in order to determine the rate of 30delG mutation, we screened for the latter, by a PSDM method (PCR-site directed mutagenesis), 300 unrelated Lebanese subjects equally distributed by sex and community for the 30delG mutation. This mutation was found with a carrier frequency of 1/43, which is lower than the carrier frequency of 1/31-35 found in white populations.6 27 28 Also, we did not find an excess in any particular ethno-religious community. Thus, we suggest that the 30delG mutation is common in our familial cases linked to DFNB1, most likely because of the highly mutable characteristic of a stretch of six identical nucleotides3 and not because of a particular ancestral mutation. Random DNA samples from 100 males and 100 females (400 chromosomes) were subsequently tested for the two novel mutations found in one family and none was found, confirming the absence of ancestral mutation in the GJB2 gene in the Lebanese population.
For the remaining families not linked to DFNB1 (64%), 12 already reported loci were implicated (table 1). Apart from DFNB1, the most frequent loci in our population were DFNB3 and DFNB9. DFNB3 was initially reported in Benkala, an Indonesian village,29and also in India.30 The haplotypes of the affected subjects from these families and ours were different. The DFNB9 locus was found in Lebanon,17 but also in Turkey31and India.32 In the Lebanese families, the same nonsense mutation, tyr703 to ter, was found in theOTOF gene,23 raising the possibility of an ancestral mutation at this locus in Lebanon. Finally, it is worth remembering that five different DFNB loci, DFNB9, 12, 13, 14, 21, and 22, were found in Lebanese families.17-22 All these results confirm the genetic diversity of the Lebanese population.
Our strategy in investigating families with NSRD has changed since the report of the mutations in the GJB2 gene. Affected patients are first tested for the 30delG mutation and the gene is sequenced if it is absent. If the results are negative, elimination of the known loci and a genome wide search is performed. In one family, several affected subjects with 30delG present on one allele were detected. No other mutation was found after sequencing. The genome wide search allowed us to link this family to DFNB12. This finding agrees with the suggested possibility6 that some subjects might be carriers of the 30delG mutation because of its high frequency in the population and have mutations leading to hearing loss in a different gene. Amazingly, this family also contained subjects with Usher syndrome, linked to the Usher 2A locus.
In conclusion, the precise number of people affected with early and profound hearing loss in Lebanon is unknown. Nevertheless, because of the high rate of consanguineous marriages, the incidence may be higher than has been reported world wide. Our findings indicate that screening for one mutation, 30delG, would allow the early detection of about one third of NSRD cases in Lebanon. Further analysis of the different genes implicated in hearing loss in the Lebanese population will improve the performance of clinicians providing genetic counselling, especially in consanguineous Lebanese families.
Acknowledgments
The first two authors contributed equally to this work. We are indebted to the participating families for their kind cooperation. Special thanks go to Professor G Lefranc for his continuous support and advice and to Dominique Weil for her help. This work was supported by grants from the Saint Joseph University for Scientific Research.