Article Text

Statistics from Altmetric.com
While searching for germline mutations inMLH1 and MSH2mismatch repair genes in patients affected with hereditary non-polyposis colorectal cancer (HNPCC), we have observed that human chromosome 3 carries two main haplotypes of the housekeeping geneMLH1. This so called caretaker gene acts as a major guardian of the genome,1 and cells in whichMLH1 is inactivated develop a characteristic mutator phenotype, as a result of a default in post-replicative DNA mismatch repair.2 3 In humans, the protein encoded byMLH1 forms at least two dimeric factors with either PMS2 orMLH3.4-6 Germline defects inMLH1 account for a number of sporadic epithelial cancers and for the majority of cases of HNPCC,7 which is the most common form of all familial cancers along with breast cancer. In addition to its role in DNA editing, the multifunctional MLH1 gene is thought to participate in mitotic and meiotic recombination,8 as well as in apoptosis.9 InE coli, evidence has accumulated to suggest that MLH1 MutL homologue may behave as a “molecular matchmaker” by acting as a chaperone, which facilitates the conformational changes required to assemble a DNA repair proficient complex from its individual components.10 In yeast, studies on segregation data of all genes known to participate in mismatch repair have shown that the MLH1gene plays a predominant role in promoting crossing over,11 and at least in mice, the MLH1 protein appears to be a component of the late replication nodules that probably prevent non-homologous genetic recombination between homeologous sequences.8 During chromosome pairing in meiosis I, the protein foci allow even better mapping of crossover events and interference distances than using chiasmata.12 Our data suggest that two major haplotypes spanning at least 55 kb of theMLH1 gene, completely specific across at least 27 kb of genomic DNA, are not functionally equivalent with respect to meiotic genetic recombination.
Materials and methods
MICROSATELLITE ANALYSIS
Genomic DNA was purified using standard phenol-chloroform extraction methods and amplified for 30-35 cycles in 20 μl (3-5 pmol of each primer, 50 μmol/l of each dNTP and 0.1 unit ofExtra-pol II DNA polymerase from Eurobio, Les Ulis, France). Primers and cycling conditions used to amplify microsatellites were as described in the Genome Database,http://gdbwww.gdb.org. The upstream primer was labelled at its 5′ end using IRD800 labelling (Lincoln, NE). A total of 0.5-1 μl of reaction product was denatured and electrophoresed on denaturing polyacrylamide gels (obtained by mixing 19 ml of 8% Sequagel XR from National Diagnostics, Atlanta, GA, with 952 μl of Long Ranger, FMC, Rockland, ME). Samples were separated and analysed on a Li-Cor 4000 automated sequencer.
SEQUENCE ANALYSIS
Sequence analysis was carried out from PCR products amplified from genomic DNA. When necessary, the PCR products were purified using a QIAquick PCR purification kit (Qiagen, Chatsworth, CA). Cycle sequencing was performed using IRD800 labelled primers (Lincoln, NE) and ThermoSequenase (Nycomed Amersham, Buckinghamshire, UK). DNA templates were sequenced in both directions and, whenever necessary, results were confirmed by sequencing two independent PCR products. Samples were separated and analysed on a Li-Cor 4000 automated sequencer. Sequences of the primers used are available upon request to PH (pierre.hutter{at}ichv.vsnet.ch).
IDENTIFICATION OF THE A/T POLYMORPHISM ATMLH1 IVS11 BYDraI DIGESTION
For the analysis of the A/T polymorphism at IVS11-7, a 178 bp fragment of genomic DNA was amplified in a final volume of 20 μl using the forward primer 5′-CTTCTTATTCTGA GTCTCTCC-3′ and the reverse primer 5′-TGGGCATAGACCTTATCACTAC-3′. The following cycling conditions were used: 94°C for four minutes, followed by six cycles of 94°C for 30 seconds, 59°C for 15 seconds (−1°C/cycle), 72°C for 30 seconds, and 26 cycles of 94°C for 30 seconds, 53°C for 15 seconds, 72°C for 30 seconds, with a final extension step at 72°C for four minutes. Then 5 μl of PCR product were digested in a 10 μl reaction using 8 units of DraI. When an A nt is present at the IVS11–7 site, the PCR product is cut into 112 bp and 66 bp products, which can be visualised on a 4% agarose gel (1.5 % Biorad, Hercules, CA and 2.5 % NuSieve, FMC, Rockland, ME).
STATISTICAL ANALYSIS
Alleles at all microsatellite markers analysed were numbered consecutively according to decreasing size. In intron 11 (IVS11) an insertion of eight nucleotides in BAT-21 is carried by allele 2 and not by allele 1. At both exon 8 and IVS14 polymorphic sites, allele 1 carries A nucleotides and allele 2 carries G nucleotides. Analysis of linkage disequilibrium was performed by permutation testing of all pairs of loci using the EM algorithm.13 Overall significance was assessed by generating 1000 random permutations of the haplotypes and p values were determined by exact tests. For each pair of polymorphic sites, chi-square tests were performed on contingency tables. Haplotype frequencies were estimated from maximum likelihood frequencies based on a total number of 4742 possible haplotypes.14
SOURCES OF THE CHROMOSOMES STUDIED
About half of the subjects analysed in this study came from 44 HNPCC or HNPCC-like families, originating from nine European countries. Nearly 30% of them carried a pathogenic germline mutation in one allele of the MLH1 gene, whereas 70% were not mutation carriers. Another quarter of the patients analysed came from a control population, the majority of Swiss origin, with a few exceptions whose nationality was not recorded. The last quarter came from nine North American families, including six large CEPH families. Thus, over 90% of the chromosomes under study were wild type with respect to mismatch repair genes.
Results
IDENTIFICATION OF TWO MAIN HAPLOTYPES AT THEMLH1 LOCUS
In the course of our search for germline mutations and polymorphisms in MLH1 in HNPCC patients, we observed that two common polymorphisms in this gene appear to be in strong linkage disequilibrium, at least in Europe and North America. The first polymorphic site lies in exon 8, where A or G nucleotides (nt) at codon 219 encode either an isoleucine or a valine amino acid. The second polymorphism also consists of either A or G, 19 nt upstream of the 3′ splice site of IVS14. In addition to our own data, we have compiled data collected by seven European centres working on HNPCC (data not shown) and a consistent pattern has emerged. At the exon 8 polymorphic site, A nucleotides appear to occur consistently in about 55% of the chromosomes analysed, a frequency which is close to that of A nucleotides (about 62%) observed at the IVS14 polymorphic site. These conserved figures prompted us to examine the possibility of linkage disequilibrium between alleles carrying two A nt and alleles carrying two G nt at the above polymorphic sites. The polymorphic status of 362 chromosomes from nine European countries and from North America was determined by DNA sequencing in chromosomes homozygous for either polymorphism as well as in heterozygous chromosomes whose polymorphic status was partly known because one homologue was tagged with a germline mutation. A simple pattern emerged, fully compatible with 211 chromosomes having two A, 114 having two G, and 37 having A and G nt, at exon 8 and IVS14 polymorphic sites, respectively. No chromosome was found to have G and A at the above respective sites.
FURTHER HAPLOTYPING OF TWO MAJORMLH1 FORMS
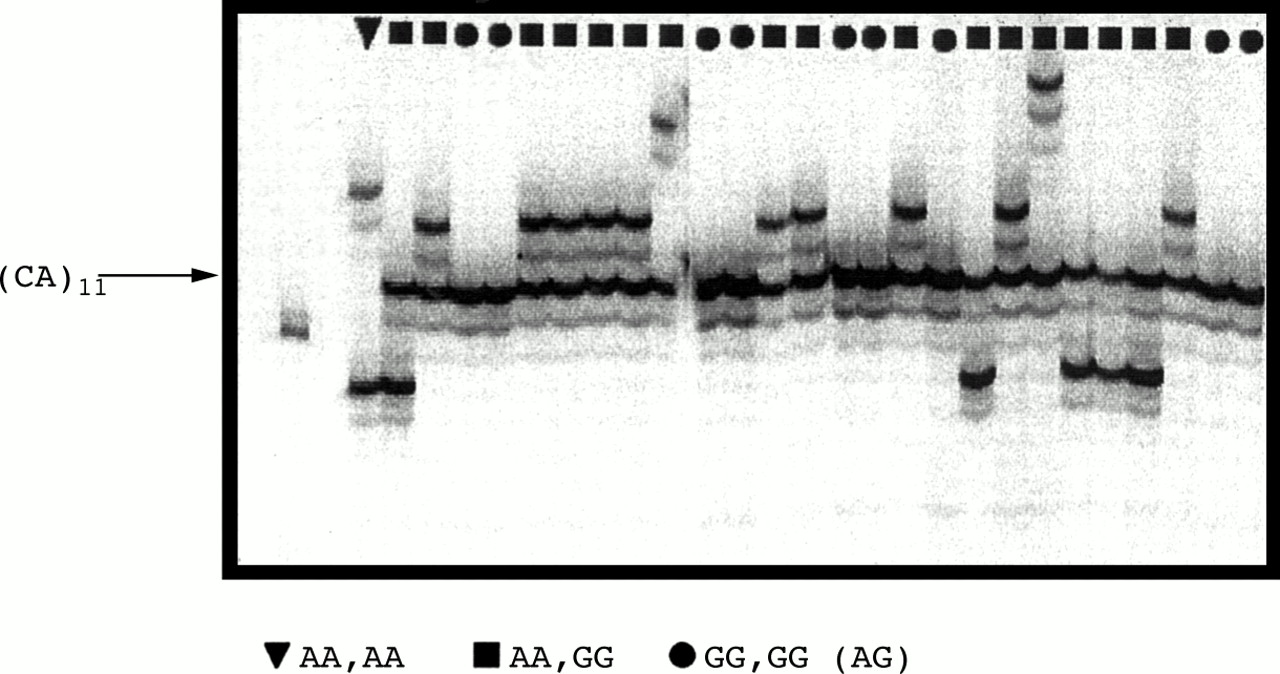
The haplotypes of the MLH1 segments extending from exon 8 to IVS14 were further characterised by analysing three additional polymorphic sites contained within a sequence of 27 kb of genomic DNA, extending from IVS11 and the above IVS14 A/G polymorphism. One of these central markers was microsatellite D3S1611 which lies in IVS12 (P Peltomäki, personal communication), and another marker corresponded to microsatellite BAT-21, which comprises a run of 11 TA dinucleotides directly followed by a run of 21 T mononucleotides, just 7 nt upstream of the acceptor splice junction of exon 12. Based on the above four intragenic polymorphic sites, the maximum likelihood haplotype frequencies were computed from a sample of 170 chromosomes and the results are shown in table 1. Only seven possible haplotypes were found. The first two haplotypes carried either allele 2 (G) or rarely allele 1 (A) nt at exon 8, allele 1 (that is, no insertion) at BAT-21, 11 CA at D3S1611, and allele 2 (G) at IVS14. In contrast to this, all other haplotypes had allele 2 at both external markers, allele 2 (insertion of 8 nt) at BAT-21, and five different allele sizes at D3S1611, ranging from 11 to 17 CA. A sixth allele (allele 3), which was later associated with the same haplotype at D3S1611, was not present in this series. Thus, a single haplotype was found for the 114 chromosomes carrying G nt at both exon 8 and IVS14, separated by about 55 kb of genomic DNA. As mentioned, this haplotype was completely shared with that of the relatively rare chromosomes (37 out of 362) which had A and G nt at the above polymorphic sites. Fig 1summarises the two main haplotypes which were found at theMLH1 locus, the commonest one associated with alleles carrying 2 A nt (hereafter referred to as A-MLH1), and the rarest one associated with alleles typically carrying 2 G nt (hereafter referred to as G-MLH1) at exon 8 and IVS14. The rare chromosomes which carry A at exon 8 and G at IVS14 are most likely to result from a historical recombination event between exon 8 and the 3′ end of IVS11, which are separated by about 28 kb of DNA. Markers BAT-21 and D3S1611 were used together with the polymorphisms at exon 8 and IVS14 for an analysis of linkage disequilibrium between all possible pairs of markers. The central part (in bold) of table 2 shows that testing of linkage disequilibrium confirmed very strong linkage disequilibrium between the four polymorphic sites, with each pair being significant at exact test p values well below 0.0001. Remarkably, the data indicated that the 151 G-MLH1chromosomes which had GG or AG nt at exon 8 and IVS14 sites, respectively, were monomorphic for 11 CA repeats at D31611, a repeat number which was not encountered among the 211 chromosomes that carried A nt at both polymorphic sites (fig 2). In contrast to this, six allele sizes were observed for A-MLH1 chromosomes at D3S1611 in our data set, ranging from 8 to 17 CA repeats, as later confirmed by direct sequencing. Similarly, all G-MLH1 alleles yielded BAT-21 products of 114 bp, with rare exceptions of 115 and 116 bp, referred to as allele 1, whereas all A-MLH1 alleles yielded BAT-21 products ranging from 120 to 124 bp (with one exception of 107 bp), referred to as allele 2. Results from sequence analysis indicated that the differences in BAT-21 length between alleles 1 and 2 correspond to a gain/loss of three TA and/or one or two T. By and large, no overlap was found between A and G chromosomes across at least 27 kb ofMLH1 genomic DNA extending from IVS11 to IVS14, and only 37 out of 362 (10.2%) chromosomes overlapped in a segment extending from beyond 27 kb to 55 kb of DNA.
Frequencies of the seven possible haplotypes at four markers within MLH1 (n=170)

Diagram showing the differences in nucleotide sequence between A-MLH1 and G-MLH1 forms.
Tests for pairwise linkage disequilibrium at four intra-MLH1 loci and four loci closely linked to MLH1
{kind=link}
{kind=link}
Allele profiles at microsatellite D3S1611 in relation to nucleotides present (AA, GG, or AG) at exon 8 and IVS14 polymorphic sites of the MLH1 gene, respectively.
BOTH A- AND G-MLH1 FORMS APPEAR TO BE ANCESTRAL
In order to extend the characterisation of the two mainMLH1 haplotypes to DNA directly flanking the gene, we took advantage of the dense microsatellite marker map described around MLH1.15 For this purpose we analysed microsatellites D3S1561, D3S1277, D3S1298, and D3S3527 located just adjacent to MLH1 on either side, and spanning a genomic region corresponding to a genetic distance of about 2 cM. Table 2 shows the results from tests for linkage disequilibrium from a series of 170 chromosomes for which the above four MLH1 flanking markers were typed, together with the four intra-MLH1 markers. Interestingly, the block of MLH1 DNA which showed strong linkage disequilibrium appears to extend to centromeric marker D3S1298, pointing to the possibility that a regulatory element might be associated with the MLH1dimorphism. The most telomeric marker, D3S1561, was in linkage disequilibrium with D3S1277, as was the most centromeric marker D3S3527 with D3S1298, consistent with the fact that these markers lie very close to each other. Indeed, in large panmictic populations, linkage disequilibrium is likely at genetic distances well below 1 cM.16
We have emphasised that the relative frequencies of A and G-MLH1 forms appear to be relatively constant at least across Europe, and our data indicated that allelic diversity at the eight markers studied is very similar between A and G-MLH1 forms. Indeed, the same allelic series were observed for each form at microsatellites linked toMLH1. Table 3 shows that the allele sets observed per microsatellite were very similar between A and G forms, with the outstanding exception at D3S1611 CA repeat. A slightly higher number of allele sizes of A alleles was expected, since A alleles are a little more frequent than G alleles in the general population, thus increasing the likelihood that rare allele sizes were represented in our relatively small sample. The similarity between allele sets found for each MLH1 form again supports founder effects, suggesting that both chromosome types have extensively recombined between MLH1 and nearby markers. When the eight markers investigated were tested from a sample of 170 chromosomes for maximum likelihood haplotype frequencies, 90 possible haplotypes were found (table 4). Again a similar haplotype diversity was associated with A and G-MLH1 forms, with nine and eight of these haplotypes occurring at frequencies above 0.02 for each form, respectively. This situation further supports an ancestral origin of both A and G forms, as it is not compatible with one allele being derived from the other in the recent past. It is worth mentioning that we have observed on two occasions a dimorphism (differing by one dinucleotide) of marker D3S1277 on A-MLH1 alleles tagged with a founding mutation among members of an extended HNPCC kindred.17
Number of allele sizes at five microsatellites linked to the MLH1 gene in relation to A-MLH1 and G-MLH1 haplotypes
Number of possible A- and G-MLH1 haplotypes with respect to eight markers, of which four are intragenic and four are closely linked to MLH1, as shown at the bottom
Discussion
We have shown that A and G-MLH1 forms are found at frequencies of about 0.6 and 0.4, respectively, throughout Europe and North America. Moreover, both forms were found to be associated with similar allelic heterozygosity at markers closely linked to MLH1. These observations strongly suggest that the two forms are ancient and have evolved separately. With regard to this, the unique size of D3S1611 in G alleles, which was not found among 211 A alleles analysed, is particularly intriguing. Historical recombinations over an interval of 60 kb are expected to occur at a rate of more than 50% in 1000 generations (approximately 25 000 years), assuming the average correspondence 1 cM=1 Mb. When this is taken into consideration, the lack of recombinants over 40-60 kb in G forms in our large sample requires an explanation. Considering the multiple functions of the MLH1 gene involved in genomic integrity, the existence of two main forms of the gene raises the possibility that these forms are not functionally equivalent. We would like to hypothesise that the difference in genetic stability between A and G forms is a consequence of a stronger avoidance of unequal crossing over by G/G and maybe by A/G genotypes, when compared to A/A genotypes. At D3S1611, this would prevent the generation of repeat sizes departing from the original 11 CA repeats in G/G genotypes, in contrast to the situation in A/A genotypes, where expansion or contraction of CA repeats may be more likely to occur. If unequal reciprocal exchange of genetic material is less likely to take place between G/G (and possibly A/G) alleles than between A/A alleles, one would expect the 11 CA repeat in G alleles to be the most stable of all the microsatellites investigated in our study. In A alleles, the most frequent repeat sizes at D3S1611 contain 13, eight, and 17 CA. According to our hypothesis, it can be argued that the sizes of eight and 17 CA have resulted from historical unequal crossovers between two ancestral chromosomes carrying 13 CA. This would imply that theMLH1 forms may be associated with different protective effects against deleterious rearrangements resulting from inter-repeat crossovers. As the human genome is loaded with dispersed repeated sequences, its integrity is constantly threatened by unequal crossing over that can occur between functional sequences which contain repeats or between homeologous sequences.
EVIDENCE FROM HISTORICAL RECOMBINATION FOR AN ASSOCIATION BETWEEN A- AND G-MLH1 FORMS AND DIFFERENT RECOMBINATION RATES
G chromosomes were associated with no sequence variation at D3S1611 and less variation at BAT-21 and nearby A/T polymorphism than G alleles (fig 1). As already mentioned, we would argue that this difference may be a consequence of unequal crossover occurring more frequently between chromosomes carrying the A-MLH1 haplotype than between chromosomes carrying the G-MLH1 haplotype, resulting in A alleles deviating more frequently than G alleles from the original phenotype. We plan to address this issue further by in vitro studies, by comparing the recombination rates between cell lines homozygous for each haplotype. Nevertheless, it should be pointed out that such an approach to experimental evolution may not necessarily reflect the actual situation that intact cells have experienced in whole organisms over long evolutionary periods. For example, if a polygenic effect is involved, it is possible that a difference in accuracy of crossing over only becomes apparent when the relevant MLH1allele is coupled with another particular allele of one of its major partner genes, such as PMS2or MLH3. Besides, it is well established that genes altering recombination rates have no direct effect on the phenotype, but alter sets of alleles at other loci with which they will be associated in future generations.18
CAN A- AND G-MLH1 ALLELES BE SUBJECTED TO DIFFERENT SELECTIVE REGIMENS?
The physical distance between the most distant markers used in our study, D3S1561 and D3S3527, is estimated to be around 2000 kb assuming that 1000 kb corresponds to 1 cM. Noticeably more meiotic recombinants were scored at markers distal rather than proximal toMLH1, and this decay of linkage disequilibrium with increasing distance adds evidence for a balanced polymorphism, in a way reminiscent of what has been described for theAdh locus inDrosophila.19 Our data are compatible with G-MLH1 alleles having diverged from an A-MLH1 allele early in human evolution, and that the two A to G transitions which occurred in G alleles did not occur later in A alleles. This may reflect a relatively lower survival rate of A alleles in the long term, again reflecting a possible consequence of enhanced alteration of genes, for instance via conversion to their inactive pseudogenes. When considering a possible selective advantage of G alleles relative to A alleles, it is worth mentioning that the T>A polymorphism which we identified at nt –7 in IVS11 (fig 1), though present in Europe, North America, and Africa (M G Dunlop, personal communication), was found in only 39 out of 548 (7.2%) A chromosomes, and that all 39 chromosomes had 13 or occasionally 14 CA repeats at the D3S1611 marker. It is also worth emphasising that MLH1 haplotype analysis from 19 HNPCC families20 (manuscript in preparation) indicated that the majority of chromosomes harbouring aMLH1 germline mutation had the G haplotype, although this haplotype is slightly less frequent in the general population than the A haplotype. Indeed, 12 out of 19 (63.2%) of theMLH1 germline mutations were found to be carried by G-MLH1 chromosomes. This observation may suggest a selective advantage of G alleles over A alleles, assuming that carriers of either allele accumulate nucleotide substitutions at similar rates. This feature could partly explain why G alleles, which may have been derived from A alleles, have at present reached a frequency close enough to that of A alleles world wide.
MAINTENANCE OF A BALANCED DIMORPHISM OF THEMLH1 GENE
We have hypothesised that G-MLH1alleles, when compared to A-MLH1 alleles, may better inhibit, at least in the long term, non-homologous recombination and thus prevent gene conversion. This could have important biological consequences, at least at minisatellite sequences, where allelic variation can result from a recombination based process and play an important role in gene expression and thus in human disease.21 22 Better prevention of non-homologous recombination by G alleles may be further enhanced if nucleotide substitutions occurring in them have a greater chance to spread in the population than when they occur in A alleles. Similarly, the early divergence between the two MLH1 alleles may have been favoured if the first nucleotide substitution(s) occurring in G alleles acted as an inhibitor of homologous recombination between the two diverging sequences or else that intermediate forms were somehow selected against.
Our hypothesis considers that a dimorphic system at theMLH1 locus may help to lead to alternative evolutionary outcomes, driving cells homozygous for A or G alleles to different levels of genetic variation upon which natural selection can act. Such a reservoir of genetic variability may be particularly critical to the fate of populations meeting challenging circumstances. Besides, one can envisage that the postulated functional difference between A and G alleles is also associated with different degrees of genomic integrity at the somatic level. Although this is clearly speculative at this stage, it would seem plausible that a slight difference in somatic rate of crossing over may influence the process of ageing as well as susceptibility to carcinogenesis.
Little can be said about the possible functional implications of the four polymorphisms which we have described withinMLH1, but it can be argued that two of these polymorphisms may not be neutral on the following grounds. The polymorphism at exon 8 changes an isoleucine into a valine, both of which are neutral and hydrophobic amino acids, but a number of cases have been reported for which the same amino acid change results in a functional difference.23 InMLH1, this polymorphism does not appear to influence predisposition to colorectal cancer, but it may not be neutral with regard to another role played by the multifunctional gene. The second sequence polymorphism which may not be functionally neutral is the 8 ± 2 nt variation near the acceptor splice site of exon 12. Indeed, the stretch of 11 TA followed by 21 T, which is present 7 nt upstream of the splice junction of exon 12, is flanked upstream by the branch point site (BPS). Interestingly, exon 12 is known to be abnormally spliced in the majority of samples from either blood or colonocytes of normal subjects,24 and skipping of this 371 bp long exon is predicted to result in disruption of theMLH1 open reading frame. Early in mammalian spliceosome assembly, U2AF65 protein binds to the pyrimidine tract between the BPS and the first AG present downstream. U2AF65 crosslinking is replaced by crosslinking of three proteins which interact in the region spanning from immediately downstream of U2 snRNP's binding site at the BPS to just beyond the 3′ splice site, showing the existence of local constraints for the catalytic step involved in the process.25 These constraints are on both the sequence and the distance between the BPS and AG, with efficient selection of the 3′ splice site depending on an optimal distance of 13 to 22 nt between the BPS and the AG.26
Linkage disequilibrium between intragenic or 5′ UTR variants has been occasionally reported for other cancer genes such asBRCA1 or CDKN2A, involved in predisposition to breast/ovarian cancer and melanoma, respectively.27 28 However, these studies were based on geographically limited sampling and the genomic distance separating the linked polymorphisms was smaller than that reported in the present study. To our knowledge, only one example reminiscent of the two discrete MLH1 haplotypes has been reported, in a region 5′ to the human δ globin gene.29 Two types of chromosomes (R and T) were found in whites, blacks, and orientals, and the factors accounting for the maintenance of this balanced polymorphism are still unknown. Our data suggest that A- and G-MLH1 alleles are sensitive targets for natural selection, but the forces underlying this process remain to be identified. Both experimental and theoretical studies have shown that selection for increased recombination can be increased in fluctuating environments.18 Diversifying selection may be partly responsible for maintaining a balanced equilibrium between the two forms, if one of them is favoured when rare, but discriminated against when it becomes more abundant. Alternatively, periodic fluctuations of the two MLH1 forms may reflect a process of genetic random walk, which can lead to either fixation or loss of alleles, depending on their degree of adaptability.
Acknowledgments
We are very grateful to L Excoffier for help with statistical analysis. We thank W Friedl, A Lindblöm, P Peltomäki, A Piepoli, P Radice, and J Wijnen for communicating information on their data on exon 8 and IVS14 polymorphisms. H Cann at the CEPH (Paris, France) provided us with DNA samples from American families. This work was supported by the following institutions to P Hutter: Recherche Suisse Contre le Cancer (AKT446), Ligues Genevoise et Valaisanne Contre le Cancer (grant 57), Fondation E Boninchi, Fondation pour la Lutte Contre le Cancer, and Fonds National Suisse de la Recherche Scientifique.