Article Text

Statistics from Altmetric.com
- ASD, atrial septal defect
- BAC, bacterial artificial chromosome
- CHD, congenital heart defects
- FISH, fluorescence in situ hybridisation
- GU, genito-urinary
- IHH, idiopathic hypogonadotropic hypogonadism
- LCR, low copy repeat
- NVDCCs, N-type voltage dependent Ca2+ channels
- OTCS, Opitz trigonocephaly C syndrome
- SNP, single nucleotide polymorphism
- SRO, shortest region of overlap
- VSD, ventricular septal defect
- contiguous gene syndrome
- deletion 9q34.3
- genotype-phenotype correlation
- Opitz trigonocephaly C syndrome
- telomere
Submicroscopic deletion del(9)(q34.3) is a rare constitutional microdeletion syndrome involving the gene-rich subtelomeric region of the long arm of chromosome 9, with about 30 cases reported.1,2,3,4,5,6,7,8,9,10,11,12 Visible constitutional 9q34 deletions are extremely rare, with only a few cases described.2,10,12 The low prevalence of large terminal deletions at the 9q34 chromosome region in liveborns is thought to reflect lethality in early embryogenesis.13
At least 18 patients with 9q34.3 microdeletions detected by fluorescence in situ hybridisation (FISH) testing, in whom normal karyotypes were initially obtained, have been reported.1,5–7,11 Many patients carry a cryptic del(9)(q34.3) that may result in a clinically recognisable phenotype characterised by severe developmental delay, mental retardation, hypotonia, congenital heart defects (CHD), seizures, and prominent craniofacial features including microcephaly, arched eyebrows, hypertelorism, short nose with anteverted nostrils, open mouth, and a protruding tongue.6,7,11 Recently, based on clinical and molecular breakpoint analyses using FISH, microsatellites, and single nucleotide polymorphism (SNP) genotyping, two research groups have independently identified an ∼1.0 Mb shortest region of overlap (SRO) of 9q34.3 deletions.6,11 Although the clinical criteria and incidence of the 9q34.3 deletion are not yet well established, the increasing number of patients reported with del(9)(q34.3) is probably related to the widespread clinical application of telomere FISH. Hence, this microdeletion syndrome may be more common than previously thought.
We have identified a 9q34.3 deletion in each of five unrelated patients, including a patient with clinical features similar to Opitz trigonocephaly C syndrome (OTCS; MIM 211750). A monogenic cause and autosomal recessive mode of inheritance have been considered probable in OTCS, usually associated with a normal karyotype.14–19 In contrast, several reports on patients with multiple congenital anomalies resembling OTCS and chromosomal imbalance of either 3p, 3q, 9p, or 11q, supported the hypothesis that OTCS is a heterogeneous condition and may be an example of an underascertained microdeletion syndrome.16,18–20
METHODS
Patients
This study was approved by the Institutional Review Board for human subjects research at Baylor College of Medicine and Affiliated Hospitals, and appropriate informed consents for children were obtained from their parents. The present research includes five unrelated children (two boys and three girls) aged from 1 to 9 years. Patients KCL1, KCL2, KCL3, KCL4, and KCL5 had an initial clinical evaluation for congenital anomalies, facial dysmorphism, hypotonia, postnatal microcephaly, unexplained mental retardation, and speech delay. They were referred for cytogenetic and subtelomere testing. The two boys were Hispanic and the three girls were of northern European descent. All subjects were born to healthy, non-consanguineous parents with no known family history of mental retardation, congenital malformation, or metabolic disorders. The clinical features are summarised in table 1 and compared with those observed in patients with a pure 9q terminal deletion as well as with the anomalies documented in reported OTCS patients.
Comparison of clinical findings observed in patients with 9q34.3 monosomy or OTCS
Key points
-
Approximately 30 individuals with 9q34.3 deletions in association with multiple congenital anomalies, recognisable facial dysmorphism, microcephaly, mental retardation, speech delay, seizures, and hypotonia, have been described recently. We report on five unrelated patients with de novo terminal 9q34.3 microdeletions, including one patient with some clinical features resembling Opitz trigonocephaly C syndrome (OTCS).
-
The deletion breakpoints were mapped using fluorescence in situ hybridisation analysis with BAC (bacterial artificial chromosome) and fosmid clones. The deletions varied in size from ∼700 kb to 2.3 Mb. The 700 kb smallest region of overlap contains two genes, EHMT1 and CACNA1B, potentially associated with the phenotypic features in 9q34.3 monosomy.
-
Specific clinical endophenotypes are correlated with the extent of the deletion, and potential candidate genes discussed. The patient with OTCS-like features has the largest deletion and the most severe phenotype among these five patients with 9q34.3 deletions.
-
Our results suggest that OTCS may be a complex phenotype due to haploinsufficiency of one or more genes within the 9q34.3 region. We propose that the 9q34.3 microdeletion represents a contiguous gene syndrome resulting in a complex phenotype depending on the extent of the deletion.
Patients KCL3, KCL4, and KCL5 had additional genetic testing for Prader-Willi, fragile X, and Rett syndromes, including MECP2 gene sequence analysis. In KCL3 and KCL4, a microdeletion of 15q12 was excluded by FISH, and a methylation assay of the SNRPN gene was found to be normal. Fragile X testing in patients KCL4 and KCL5 found their DNA to be normal, while the FISH result for Smith-Magenis syndrome in KCL4 was also normal.
Patient KCL1
This boy (fig 1A) presented with multiple congenital anomalies. Ultrasound showed both gonads in the inguinal canal, poor cortical medullary differentiation, and renal cysts. Both cranial CT and MRI documented the small size of the brain and hypoplasia of the inferior cerebellar vermis. Neuromotor development was significantly delayed, with poor head control. Two of the authors (FS and JGL) diagnosed the patient with an apparent OTCS-like phenotype. The respiratory difficulties triggered by viral illness worsened in the subsequent months and the child succumbed to sudden respiratory failure, refractory to treatment, at 26 months of age.
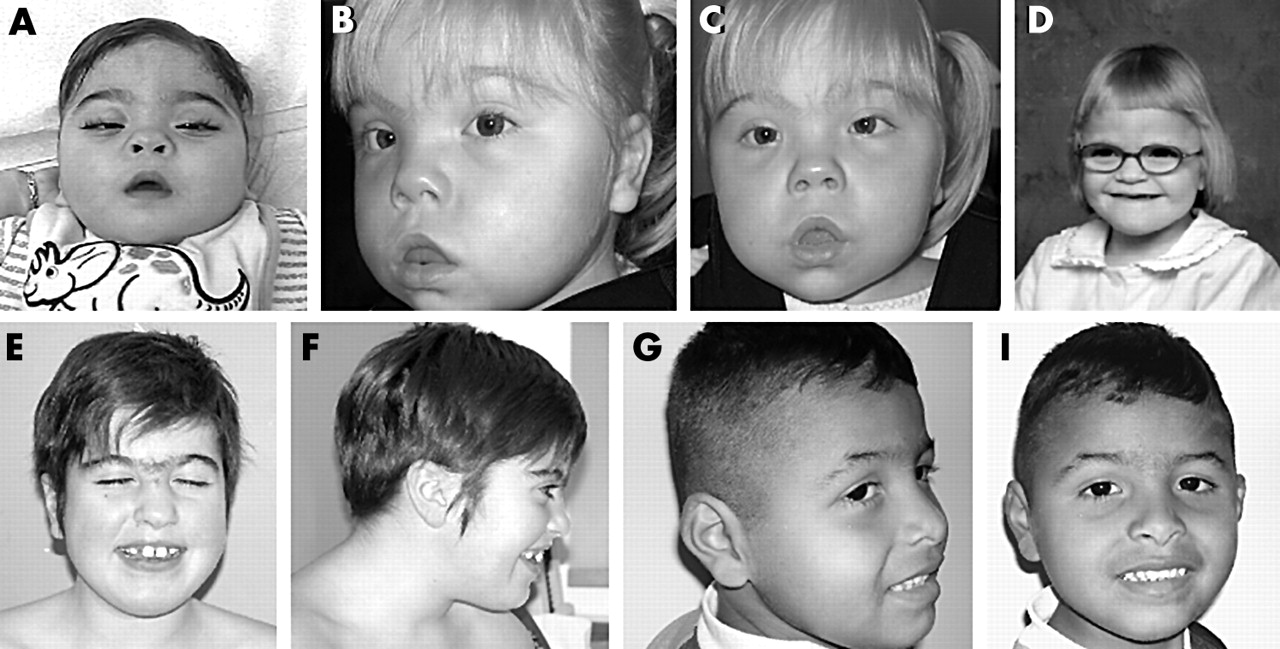
Facial photographs of the five subjects with 9q34.3 deletion. (A) Patient KCL1 at 1.5 years of age. (B, C) KCL2 at 3 years of age. (D) KCL3 at 4 years of age. (E, F) KCL4 at 9 years of age. (G, I) KCL5 at 4 years of age. Of note are midfacial hypoplasia, hypertelorism, broad depressed nasal bridge, small nose with upturned nares, arched eyebrows, synophrys, open “carp-like” mouth, large protruding tongue, and widely spaced teeth. (Photographs reproduced with permission)
Patient KCL2
This girl (fig 1B and C) was born at 32 weeks gestation by C-section and suffered respiratory distress at birth. Patent ductus arteriosus was confirmed echocardiographically but closed spontaneously. Neurological examination at 2 years of age exhibited normal muscle strength, but clonus and extensor plantar reflex in the lower extremities. Despite adequate movement in all extremities, she still had poor head control and was unable to roll over. She had no meaningful words. Brain MRI showed moderately enlarged cerebral ventricles and prominent frontal and temporal subarachnoid spaces bilaterally. There was significant hypomyelination of the periventricular white matter and centrum semi ovale, and a hypoplastic corpus callosum.
Patient KCL3
This girl (fig 1D) was born at full term following an uncomplicated pregnancy and normal vaginal delivery. She began rolling over at 5 months of age and neuromotor development was normal to 1 year of age. Neurological examination at 3 years of age revealed an immature pincer grasp and difficulties coordinating hand movements with preserved muscle strength and normal deep tendon reflexes. Developmentally, she was able to sit independently, pull to stand, and cruise. The gait was wide-based. There was an unusual distribution of subcutaneous fat across the shoulders. Brain MRI was abnormal only for subtle delay of myelination. The patient having been enrolled in a speech therapy program at 3 years of age, had learned elements of sign language by 3 years 9 months, communicated by putting two or three signs together, and recognised body parts, colours, animals, and numbers.
Patient KCL4
This girl (fig 1E and F) was born at term by induced but normal delivery after an uncomplicated pregnancy, but with concern about lack of fetal movements in the third trimester. She had no head control until 5–6 months of age and started crawling at 16 months. With her significant developmental delay, she was quite clumsy and non-verbal, but used sign language. Psychometric assessment at 4 years of age placed her at the functional level of a 26 month old child. By age 9½ years, she had developed some speech, but was still significantly delayed. Her behaviour had deteriorated and she was aggressive to family members. Picking and biting her fingernails and cuticles sometimes led to bleeding.
Patient KCL5
This boy (fig 1G and I) was born at 42 weeks gestation. Despite early intervention, his gross and fine motor development have been delayed. He began to roll over at 7 months, sat unsupported at 15 months, and walked at 2 years of age. At 3 years of age he was able to pick up small objects with a pincer-type grasp. At 4 years he was able to run, feed himself with the hands, and drink from a cup. He was unable to dress or undress himself. His speech development only consisted of a vocabulary of three words. He had no notion of either body parts or colours, and could not identify objects in picture books or videos.
Cytogenetic and FISH analyses
Cytogenetic analyses of peripheral blood were performed using standard techniques. GTG banded metaphase chromosomes were analysed at the 500–750 band resolution. The FISH assays using the commercially available 41 subtelomere specific probes were carried out on metaphase chromosomes as specified by the manufacturer (Vysis, Downers Grove, IL). A total of 18 BAC (bacterial artificial chromosomes) clones from RPCI-11 and RPCI-13 human libraries, representing a 3.4 Mb telomeric region of the long arm of chromosome 9, were selected from UCSC Genome browser (http://genome.ucsc.edu) and NCBI (http://www.ncbi.nlm.nih.gov) databases (fig 2). Two fosmid clones G248P80328A9 (enclosed ZMYND19 gene) and G248P88772F5 (enclosed ARRDC1 gene) within RP11-48C7 were selected to characterise the breakpoint of the smallest deletion. BAC and fosmid DNAs were isolated using the PSIψClone BAC DNA Kit (Princeton Separations, Adelphia, NJ). FISH probes were directly labelled with either Spectrum Orange-dUTP or Spectrum Green-dUTP using a commercially available kit (Vysis).

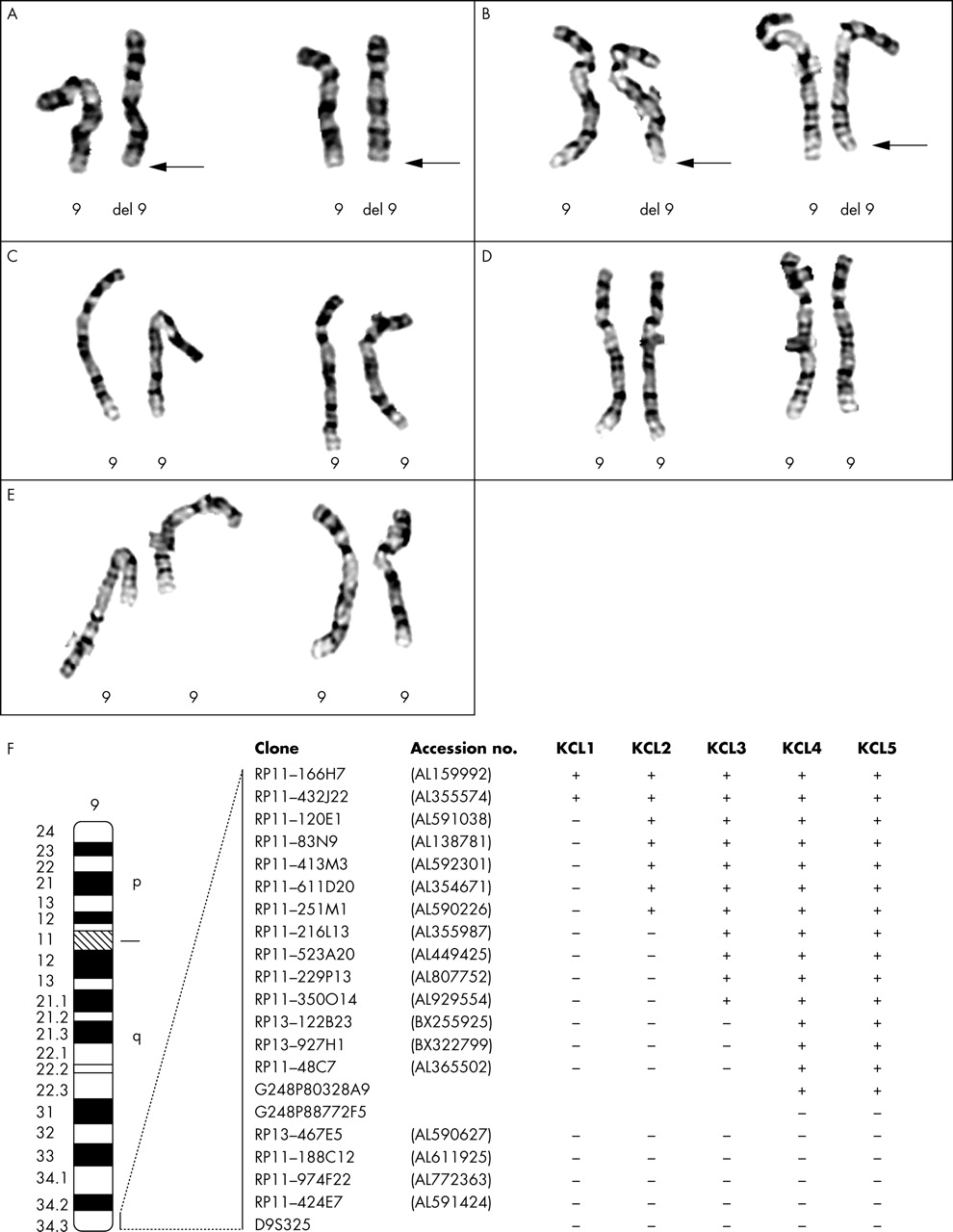
Partial high resolution G banded karyotypes and ideogram of chromosome 9. (A–E) Partial karyotypes of patients KCL1, KCL2, KCL3, KCL4, and KCL5, respectively. Arrows indicate deleted chromosome 9. (F) Ideogram showing results of the deletion mapping by FISH analysis. +: Signal present on both chromosomes 9, −: signal absent on deleted chromosome 9.
Bioinformatics and sequence analysis
We performed computer analysis of the DNA sequence of the most distal 3 Mb in 9q34.3 obtained from the UCSC Genome browser (http://genome.ucsc.edu, accessed May 2004) and, to examine for low copy repeats (LCR), compared the genome sequence against itself using the Blast 2 browser (http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html). The analysis for LCR has considered only the intrachromosomal segmental duplications in the chromosome 9q34.3 region.
RESULTS
Cytogenetic and FISH analyses
G banding analyses showed apparently normal karyotypes in all five patients (fig 2A–E). FISH for all chromosome specific telomeric regions revealed ish del(9)(q34.3)(D9S325-), consistent with a subtelomeric deletion of 9q in each patient, and normal hybridisation patterns for all other subtelomeres. FISH analyses with 9q region specific probes showed normal parental chromosomes in the KCL1, KCL3, KCL4, and KCL5 families. The parents of patient KCL2 were not available. Although all patients were initially reported as cytogenetically normal, a del(9)(q34.3) was suspected in two subjects (KCL1 and KCL2), parenthetically those with the largest deletions, following retrospective review of the GTG banded karyotype at the 750 band level (fig 2A and B). More detailed FISH investigations were helpful in determining the deletion sizes in each patient (fig 2F). The terminal deletions had different breakpoints in the 9q34.3 region ranging in size from ∼700 kb to 2.3 Mb (table 1). Patient KCL1 was found to have the largest deletion as the proximal breakpoint mapped within two overlapping clones RP11-432J22 (not deleted) and RP11-120E1 (deleted). The deletions in patients KCL2 and KCL3 were of intermediate size. The most distal breakpoint (cases KCL4 and KCL5) was found to be located between two overlapping clones RP11-48C7 (not deleted) and RP13-467E5 (deleted), hence defining the smallest deletion. We implemented fine mapping in KCL4 and KCL5 with additional fosmid clones and precisely mapped the 9q34.3 region of haploinsufficiency. The deletion is similar in size for both patients, the proximal breakpoint located between probes G248P80328A9 (not deleted) and G248P88772F5 (deleted) (fig 2F). Based on the physical distance of G248P80328A9 from the 9q telomere, we defined the smallest 9q34.3 subtelomeric deletion to a region of ∼700 kb proximally flanked by the ARRCD1 gene (fig 3).

{kind=link}
{kind=link}
{kind=link}
(A) Physical map of the 9q34.3 region (top) including known markers and their megabase position. The microsatellite markers are ordered approximately to scale according to their physical location in the contig. Horizontal lines show respective locations of the FISH probes (below). Filled black dots above clone 48C7 represent fosmid clones. (B) Schematic representation of the 9q34.3 deletions in 15 patients determined by FISH and genotype analyses. Five individuals (KCL1, KCL2, KCL3, KCL4, KCL5) were characterised by FISH in the present study. Clinical and molecular data on 10 informative patients reported elsewhere include patients P2, P5–9, P12,11 and cases B, C, and E.6 The left part of the figure shows the extent of deletion in each patient. Non-deleted chromosome regions are represented by filled horizontal bars. The hatched bars identify either uninformative or inconclusive areas. The deleted regions in each patient are indicated by a double dashed line. On the right side of the figure the clinical features in the patients are given according to the initial letters of anomaly (Trigonocephaly, Genitourinary anomaly, Brain anomaly, Epilepsy or seizures, congenital Vascular and cardiac anomalies, Laxity of joints, Infection susceptibility, Craniofacial dysmorphism, Hypotonia, Obesity, Microcephaly, Speech impairment). +: feature present; −: feature absent; blank: feature not reported or not verified. (C) Phenotypic differences correlated with 9q34.3 deletion size. Open rectangles represent the proposed position of critical regions for individual clinical findings. The critical intervals defined are based on the subject with the particular phenotypic feature and the smallest deletion. Transcribed genes in the critical interval are represented by arrows; EMHT1 is represented by a broken arrow to indicate alternative splicing.
Genotype-phenotype correlations
Detailed molecular and clinical studies were performed in our five 9q34.3 deletion patients in order to explore the genotype-phenotype correlation. Comparison of 15 subjects, consisting of ten informative patients reported in the literature and the five described herein, enabled us to construct a deletion map and to delineate 9q34.3 minimal critical regions containing potential candidate genes for the distinctive clinical features observed in monosomic patients (fig 3). We found that the association of recognisable and characteristic craniofacial features, with hypotonia, microcephaly, and global developmental delay, was present in all five of our patients and consistently found in those reported previously, regardless of deletion size. Based on the smallest deletion in our patients KCL4 and KCL5, we defined a minimal critical region of ∼700 kb which is deleted in all patients (fig 3). There are at least five obese patients with del(9)(q34.3) described in the literature,3,4,8 in addition to our patients KCL4 and KCL5. The food seeking behaviour and obesity were noted in a few cases as early as 2 years of age. The frequency of obese children in this microdeletion syndrome could be underestimated due to limited clinical description or the young age of the patients. Our results suggest that the size of the 9q34.3 deletion may be important as well. Therefore, we suggest that haploinsufficiency of a gene(s) located in the minimal critical region is responsible for the common clinical findings of craniofacial dysmorphic features, hypotonia, obesity, relative microcephaly, and delay or absence of expressive speech observed in 9q34.3 monosomy.
Other manifestations such as cardiac defects, seizures, limb and brain anomalies, episodes of recurrent respiratory infections, hypothyroidism, and abnormal genitalia are detected most commonly in patients with larger deletions. For those individuals in whom an adequate clinical description was available, six critical intervals may be delineated for specific clinical manifestations (fig 3C).
Four individuals who carry a deletion over 1.6 Mb in size (case B, case C, P12, KCL1) presented with congenital anomalies including complicated CHD, anal atresia, kidney defects, abnormal pulmonary circulation, severe growth, and psychomotor retardation. Furthermore, patient KCL1, who had the largest deletion reported in the present study, exhibited trigonocephaly and a palate with a deep furrow in addition to CHD, abnormal genitalia, and capillary haemangioma, features suggestive of OTCS. While the patients over 4 years of age with small deletions showed a tendency to accelerated growth and progressive obesity, patients with the large deletion were extremely small. The various types of monosomy 9q34.3 appear to cause a recognisable complex syndrome of multiple congenital anomalies and mental retardation due to various degrees of haploinsufficiency of physically contiguous genes. Phenotypic differences between patients correlate clearly with deletion size.
DISCUSSION
In this study, we have characterised further the 9q34.3 microdeletion syndrome, a moderately distinct and recognisable clinical entity. Based on our results, we were able to delimit the ∼700 kb critical region distal to the ARRCD1 gene, as the smallest deletion associated with the del(9)(q34.3) phenotype (fig 3). Interestingly, there are only two known genes, CACNA1B and EHMT1, located in the SRO, both highly expressed in brain. The CACNA1B (α1βCav2.2; MIM 601012) gene encodes the α1β subunit specific for the N-type voltage dependent Ca2+ channels (NVDCCs). NVDCCs are predominantly localised to the nervous system and are critically involved in neurotransmitter release.24,25 Selective block of the NVDCCs demonstrated their significant role in the migration of immature neurons,26 whereas knockout mice deficient for α1βCav2.2 provide evidence that the function of sympathetic neurons is predominantly regulated by NVDCCs and cannot be compensated for by other types of voltage dependent Ca2+ channels.27 Thus, NVDCC deficiency may result in neurological dysfunction seen in the patients hemizygous for CACNA1B. Human microcephaly is caused by a failure of normal brain growth as a result of defects in pattern formation, cell proliferation, survival, differentiation, and/or cell growth.28,29 However, observed craniofacial changes in the majority of individuals with 9q34.3 deletions cannot be easily explained by isolated haploinsufficiency of only CACNA1B.
EHMT1 (euchromatic histone methyltransferase; MIM 607001), a widely expressed gene, is involved in chromatin modification and is known to cause transcriptional silencing of genes. The particular role of EHMT1 in the molecular mechanism of epigenetic transcriptional repression remains elusive. Interestingly, different authors have noted phenotypic similarity between 9q34.3 deletion subjects and patients with Prader-Willi or Rett syndrome. In particular, three patients in our study were referred for genetic testing for both syndromes. There is a possibility that EHMT1 is involved in the same or similar biological process as SNRPN and MECP2. Remarkably, disruption of EHMT1 was identified in a child with features of 9q deletion syndrome who carries a de novo balanced translocation t(X;9)(p11.23;q34.3).30,31 These findings provide additional evidence that EHMT1 haploinsufficiency may be responsible for the del(9)(q34.3) phenotype. At the present time, we hypothesise that both genes CACNA1B and EHMT1 are dosage-sensitive and contribute to 9q34.3 deletion syndrome.
We have mapped 9q34.3 deletions in five patients and constructed a physical map of the deletions. The map encompasses 3 Mb of the distal 9q34.3 chromosomal region. Breakpoint analysis in 15 patients, for whom molecular studies were performed, did not reveal any obvious “hotspots” for recurrent chromosome breakage in this genomic interval (fig 3). A common 400 kb breakpoint interval had been proposed previously based mostly on genotyping.11 Only two of our patients have the breakpoint within the RP11-48C7 BAC clone, but seven others display a wide range of deletion sizes. For six individuals reported in the literature (P2, 5–9)11 fine mapping studies were not performed. Additionally, the bioinformatic sequence analysis did not reveal intrachromosomal LCR or segmental duplications that could potentially predispose to rearrangements.
Accurate assessment of the deletion size and the definition of precise breakpoint sequences enabled us to evaluate genes within the deleted region that may contribute to the phenotype of the del(9)(q34.3) patients. Although the interpretation is limited by the number of samples studied, our analysis uncovered a correlation between the severity of the phenotype and the size of the deletion.
Susceptibility to upper respiratory and ear infections, one of the frequent findings among 9q34.3 deletion patients, may be explained by aspiration due to swallowing problems and hypotonia, or to immune deficiency. Interestingly, four subjects (KCL3, KCL4, KCL5, case E) with deletions less than 1.4 Mb had persistent episodes of aspiration, but had no history of infection. In contrast to those with small deletions, patients with deletions larger than 1.4 Mb had prenatal growth retardation and frequent episodes of infection. The genes identified within this region include TRAF2 (MIM 601895), a receptor associated factor 2 of tumour necrosis factor. Traf2−/− mice appeared normal at birth but show postnatal growth delay and die prematurely. Atrophy of the thymus and spleen also has been observed.32 Additional immunological studies are necessary to determine whether these patients truly have immune deficiency.
Another potentially interesting gene was deleted in at least two male patients with abnormal genitalia and a female patient (KCL3) with anosmia. NELF (MIM 608137) has been proposed as a candidate gene for the autosomal form of Kallmann syndrome (MIM 147950), with key clinical components of idiopathic hypogonadotropic hypogonadism (IHH; MIM 146110) and an- or hyposmia. Mutation of NELF was found in one out of 65 patients with IHH, but not in 100 normal control individuals.33 Study of additional male individuals with deletion encompassing NELF is required to better characterise its haploinsufficiency.
The most severe phenotype was observed in the patients with large (>1.6 Mb) terminal deletions extending proximally beyond NOTCH1 (MIM 190198). The patient with the largest deletion reported in our study had some OTCS manifestations. Due to phenotypic overlap, Bohring-Opitz (C-like syndrome; MIM 605039) and Baraitser-Winter (MIM 243310) syndromes were also considered and ruled out in the differential diagnosis.
The characteristic OTCS findings have not been reported in three other individuals with large deletions (fig 3), nor in patients with cytogenetically visible 9q deletions.2,10 As OTCS is considered to be an autosomal recessive disorder,15,17 the possibility exists that our patient with 9q34.3 deletion is a phenocopy or thus affected due to the unmasking of a recessive allele on the non-deleted chromosome or to the position effect of an as yet unknown gene. We hypothesise that an OTCS causative gene(s) may be located within the 9q34.3 region, and propose OTCS or OTCS-like patients should be screened for 9q34.3 microdeletions. In any event, large 9q34.3 deletions encompassing the 2.3 Mb subtelomeric region result in a very abnormal and complex phenotype with considerably reduced life expectancy. Detailed study of additional patients with the variable 9q34.3 breakpoints will likely provide further insight into the genotype-phenotype correlation and contribute to elucidation of dosage-sensitive genes.
In conclusion, our studies have reduced the size of the 9q34.3 critical region initially reported by Harada et al6 and Stewart et al11 from ∼1.0 Mb to ∼700 kb distal to the ARRDC1 gene. These deletions encompassing the CACNA1B and EHMT1 genes appear to be correlated with the 9q34.3 microdeletion disorder. The extent of the deletions in the 9q34.3 contiguous gene syndrome reveals potential relevant genes for specific phenotypes. We hypothesise that alterations of a gene(s) located within 9q34.3 may be responsible for some cases of OTCS. Further studies will better clarify the function and the phenotypic consequences of haploinsufficiency for specific genes critical to the 9q34.3 deletion syndromes.
Acknowledgments
We thank the families for their cooperation in this research.
REFERENCES
Footnotes
-
This study was supported in part by grants from the National Institute of Child Health and Human Development (PO1 HD39420) and the Mental Retardation Research Center (HD24064).
-
Competing interests: none declared