Article Text

Statistics from Altmetric.com
Wilms tumour (WT) is an embryonal tumour of the kidney that occurs in 1 in 10 000 children. Familial clusters are rare and account for only 1–3% of cases. Mutations in WT1 account for a minority of WT families and two autosomal dominant familial WT predisposition genes have been mapped to chromosomes 17q21 and 19q13.1–3 However, a considerable proportion of familial WT pedigrees are not attributable to any of these loci.4
Fanconi anaemia (FA, MIM 227650) is a rare autosomal recessive condition affecting ∼1 in 300 000 children. FA is characterised by variable congenital abnormalities, short stature, bone marrow failure, hypersensitivity to DNA crosslinking agents, and a predisposition to haematological malignancies such as acute myeloid leukaemia in childhood.5 FA is heterogeneous and consists of at least 11 complementation groups, A, B, C, D1, D2, E, F, G, I, J, and L.6–8 Eight FA genes have been cloned and at least six FA proteins, FANCA, FANCC, FANCE, FANCF, FANCG, and FANCL, form a nuclear complex required for monoubiquitination of FANCD2. This modification promotes translocation of FANCD2 to DNA repair foci that also contain BRCA1, BRCA2, and RAD51.9
In 2002, Howlett and colleagues reported biallelic BRCA2 mutations in individuals with Fanconi anaemia D1 (FA-D1). Subsequently, additional FA-D1 and unclassified FA cases were examined and cases with BRCA2 mutations and WT and/or brain tumours were reported.10–13 These data prompted us to investigate BRCA2 in a familial WT pedigree, WILMS2, which includes siblings with both WT and brain tumours.4,14,15
METHODS
Family report
WILMS2 was ascertained as part of our research on susceptibility to WT, which is approved by the London Multicentre Research Ethics Committee. The family includes two affected brothers with WT. The elder child first came to attention when his cryptorchidism was corrected at 2 years of age. At that time hypo- and hyperpigmented areas and a few café au lait spots were noted and he was below the 3rd centile for weight, height, and head circumference. At 3.5 years of age a stage III WT was surgically removed and the child made an uneventful recovery after radiotherapy and chemotherapy with vincristine and actinomycin D. At 9 years of age he developed seizures and was found to have two intracerebral lesions, which were diagnosed as glioblastoma multiforme on stereotactic biopsy. Due to extensive disease no further surgery was attempted and the child died 13 months later. The second son had an uneventful birth and early infancy with normal height and weight, though he was noted to have three café au lait spots and many small depigmented and hyperpigmented spots. At 7 months a stage 1 WT was diagnosed and surgically removed and he was given actinomycin D without radiotherapy. Then 5 years later, whilst asymptomatic, an MRI brain scan was performed as a screening procedure in view of the family history. A cerebellar lesion was identified and resected. Histology confirmed a grade IV medulloblastoma which was treated with radiotherapy. At 10 years of age he developed pre B cell acute lymphoblastic leukaemia and spontaneous breakages of the chromosomes was noted. He was treated with chemotherapy and suffered a cerebral haemorrhage due to asparaginase. He subsequently died at 12 years of age from diffuse relapse of the medulloblastoma.
Key points
-
We present an unusual pedigree in which two brothers both developed Wilms tumour and brain tumours.
-
Two truncating BRCA2 mutations, 886delGT and S1882X, were present in each child. Biallelic BRCA2 mutations cause Fanconi anaemia D1 (FA-D1), but neither child had the typical clinical features of Fanconi anaemia.
-
Heterozygote BRCA2 mutation carriers are at risk of breast and other cancers. At presentation there were no cases of cancer in first or second degree relatives. However, after the children died their mother and paternal aunt developed breast cancer.
-
Previous reports proposed that biallelic BRCA2 mutations carriers would likely harbour at least one missense variant or truncation restricted to the carboxy terminus of the protein. However, we show there is no overall bias in mutation position in biallelic cases towards the 3′ end of the gene compared with heterozygous BRCA2 mutation carriers identified from breast cancer pedigrees.
-
We also show that BRCA2 K3326X, which is present in 1% of the population and is not associated with breast cancer predisposition, does not predispose to FA-D1, in contrast to previous suggestions.
-
Our data suggest that similar BRCA2 mutations predispose to breast cancer when monoallelic and Fanconi anaemia when biallelic. Caution in attributing pathogenicity to missense BRCA2 variants in biallelic cases should be exercised, unless they are known to predispose to breast cancer in monoallelic cases.
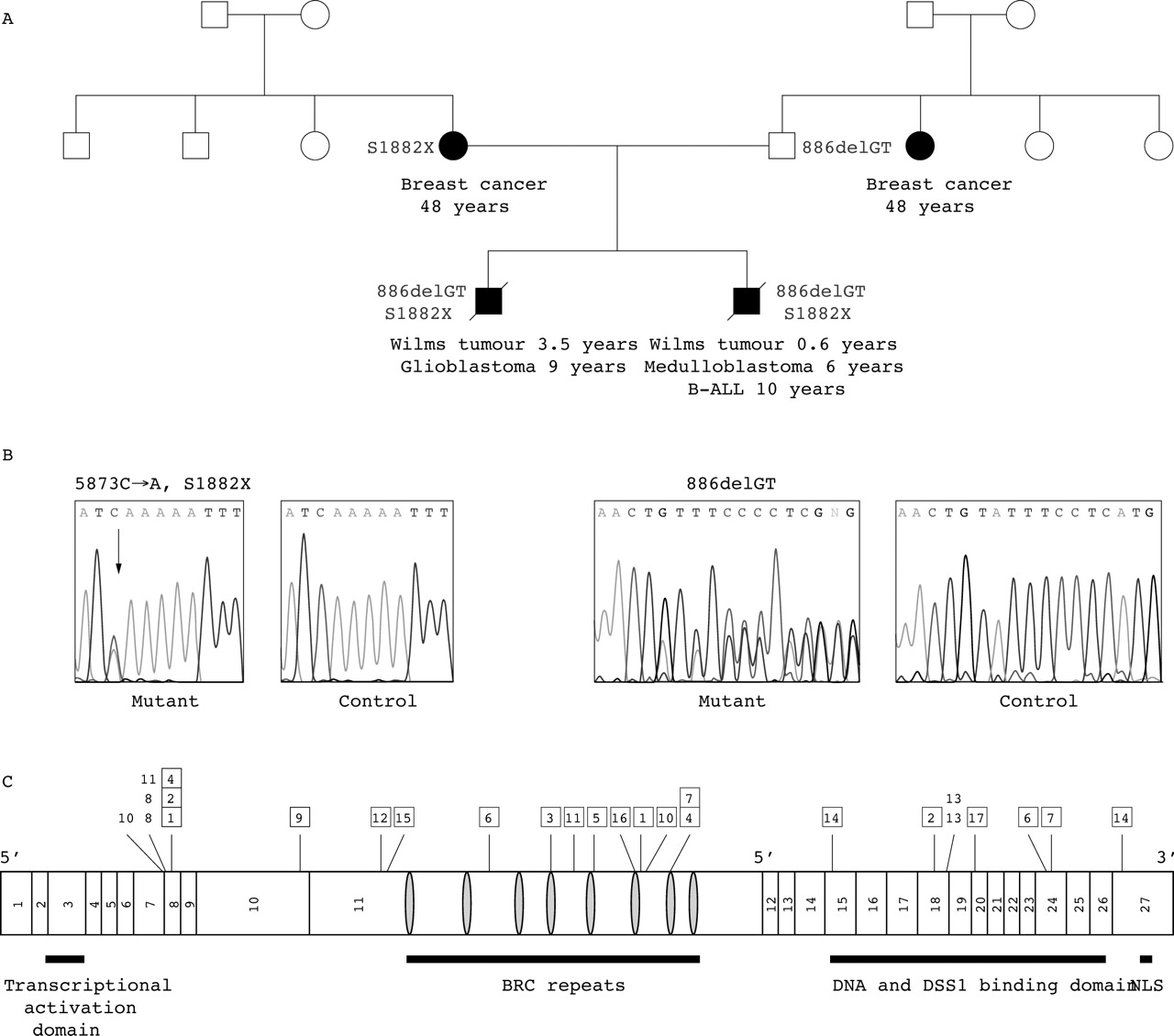
No first or second degree relative had cancer when the family presented. However, after the children had died the mother developed breast cancer at 45 years of age and a paternal aunt developed breast cancer at 48 years of age (fig 1A).

{kind=link}
Pedigree and BRCA2 mutations in WILMS2 and position of all known mutations identified in biallelic BRCA2 cases. (A) Pedigree structure of WILMS2; closed symbol: affected with cancer, open symbol: unaffected with cancer. The type of cancer and age at presentation are given under the symbol. (B) BRCA2 mutations identified in WILMS2 and wild type sequence from a control. (C) Position of mutations identified in all reported cases with biallelic BRCA2 mutations. The exons and positions of functional domains in BRCA2 are shown. NLS, nuclear localisation signal. The numbers along the top refer to the family numbers in table 1. Boxes around family numbers indicate frameshift or nonsense mutations, while family numbers without a box indicate splice site mutations. Variants of unknown significance and polymorphisms are not shown.
Laboratory analyses
We screened genomic DNA from the two affected brothers through the complete coding sequence of BRCA2 by a combination of conformation sensitive gel electrophoresis16 and direct sequencing using the BigDye Terminator Cycle Sequencing Kit and an ABI 3100 automated sequencer. BRCA2 primer sequences and conditions are available on request.
To evaluate the K3326X and N372H variants we screened 148 BRCA2 mutation positive cases from 69 families ascertained from the Breast Cancer Susceptibility Collaboration (UK) using the ABI Prism 7900HT Sequence Detection System (TaqMan; Applied Biosystems, Foster City, CA). TaqMan primers and probes were designed by Applied Biosystems SNP Genotyping Assays-By-Design service (sequences available on request). Assays (12.5 μl) were performed on 10 ng genomic DNA with 1×TaqMan Universal PCR Master Mix and the Assays-By-Demand standard thermal cycler protocol. Plates were read on the ABI Prism 7900HT Sequence Detection System in end point mode using the allelic discrimination Sequence Detection software (Applied Biosystems). Heterozygotes were confirmed by direct sequencing using a fresh template.
RESULTS AND DISCUSSION
Mutational screening of BRCA2 in the two affected brothers revealed two truncating mutations in each child (fig 1B). The paternally inherited mutation was a deletion in exon 8, 886delTG, which is predicted to truncate the protein at codon 223, before the eight BRC repeats. The maternally inherited nonsense mutation in exon 11, S1882X, is predicted to truncate the protein such that BRC7 and BRC8 would be absent (fig 1C).
Sixteen other families with biallelic BRCA2 mutations have been reported, mainly from investigation of unassigned FA cases (table 1, fig 1C).10–13 Our findings broaden the spectrum of phenotypes associated with biallelic BRCA2 mutations to familial WT pedigrees and cases without a pre-existing diagnosis of FA. Although a diagnosis of FA had been made in the other biallelic BRCA2 families, the clinical features were not always typical and similarities with WILMS2 are apparent. Overall, biallelic BRCA2 mutations appear to be associated with unusually high spontaneous chromosome aberration rates, less frequent bone marrow suppression, less frequent skeletal abnormalities, and a different spectrum of childhood cancers compared with other FA subtypes.10–13 WT (five cases), brain tumours (nine cases including medulloblastoma, glioblastoma multiforme, and astrocytoma), acute myeloid leukaemia (seven cases), and acute lymphoblastic leukaemia (three cases) have been identified in 23 biallelic BRCA2 mutation carriers. Of these cases, 21 have had childhood cancer and in four cases multiple cancers occurred (table 1). Only one case has reached adulthood without cancer and he is homozygous for an in-frame deletion of four codons and thus may have a milder phenotype.10 Altogether, although there has been some bias towards screening childhood cancer cases, the available data suggest the cancer risk of children with biallelic BRCA2 mutations may be very high.
Mutations and cancers in biallelic BRCA2 mutation cases
The early identification of the BRCA2 status of these children is important, as it will likely affect response to cancer therapies. In particular, DNA damaging chemotherapeutic agents may result in severe toxicity at standard doses. The use of radiotherapy will also need careful consideration. The brothers in WILMS2 both received radiation treatment without obvious toxicity. This is consistent with recent evidence that abrogation of BRCA2 function has little effect on radiation sensitivity unless the p53 dependent DNA damage checkpoint is also inactivated.17 However, irradiation of murine cells homozygous for an exon 11 BRCA2 truncation has been shown to substantially increase the spontaneous somatic mutation rate compared with wild type animals.18 If this increase in somatic mutation acquisition also occurs in humans, it would suggest that radiotherapy treatment of biallelic BRCA2 cases might increase the risk of second or third cancers.
The initial report from Howlett and colleagues hypothesised that FA-D1 would occur in cases with at least one BRCA2 mutation that was less severe, for example, a missense alteration, splicing defect, or truncation in the carboxy terminus of the gene. It is of considerable importance to clarify whether such variants confer a risk of FA, given their prevalence and the implications for genetic counselling. In five reported pedigrees (families 3, 9, 12, 16, and 17; table 1) a missense variant was proposed as one of the FA predisposing mutations. In a further pedigree, family 15, the second mutation was reported as K3326X, which is present in ∼1% of the population and a similar proportion of breast cancer cases, and is not associated with high penetrance breast cancer susceptibility.19 Of these variants, perhaps the most plausible FA predisposing alleles are K3326X, which results in loss of 93 amino acids, and N372H, which is postulated to be a low penetrance breast cancer susceptibility allele as HH homozygotes are reported to have a relative risk of breast cancer of 1.3.20 To evaluate these two variants we screened 148 BRCA2 mutation positive cases from 69 families ascertained from the Breast Cancer Susceptibility Collaboration (UK).
Nine BRCA2 families carried K3326X. In seven unrelated families K3326X segregated with BRCA2 6503delTT, confirming the previous report that this mutation originated on a K3326X haplotype and that the two BRCA2 sequence alterations are in cis.19 In the remaining two families K3326X did not segregate with the mutations, 3386delTAGA and 6819delTG, indicating that the variant was on separate chromosomes from the mutations. Three individuals from these two families were carriers of a disease causing BRCA2 mutation and K3226X. None had evidence of FA or childhood cancer although all three developed breast cancer between the ages of 45 and 50, consistent with heterozygosity for the BRCA2 mutation. For N372H, we identified 13 individuals who carried a BRCA2 mutation and were homozygous for histidine at codon 372. None had evidence of FA or childhood cancer. These data strongly suggest that neither K3226X nor N372H are FA predisposing alleles and that caution in attributing pathogenicity to unclassified BRCA2 sequence variants should be exercised. It is likely that a second BRCA2 mutation is present in families 3, 5, 9, 12, 15, 16, and 17 but has not been identified due to insensitivity of mutation screening, as previously suggested by Offit et al.11 The inability to detect an appreciable proportion of BRCA2 mutations by standard mutation detection techniques has been demonstrated in breast cancer pedigrees with strong linkage to BRCA2 but no identifiable mutation.21
These data also call into question the hypothesis that biallelic BRCA2 cases are more likely to carry truncations in the carboxy terminus of the protein. To evaluate this we compared the position of truncating (frameshift or nonsense) mutations identified in FA cases with the mutation position in heterozygous BRCA2 carriers identified from breast cancer pedigrees. Eighteen separate truncating mutations have been identified in 17 independently ascertained FA families (table 1, fig 1C). We used the Breast Cancer Information Core database (http://research.nhgri.nib.gov/bic/) to identify truncating mutations detected by full gene sequencing in familial breast cancer pedigrees. A total of 507 different frameshift or nonsense mutations have been reported in breast cancer families. In biallelic BRCA2 cases, 6/18 (33%) truncating mutations occurred after exon 11. By comparison, in heterozygous BRCA2 cases, 214/507 (42%) truncating mutations occurred after exon 11. These data do not support a bias in mutation position in biallelic BRCA2 cases towards the carboxy terminus.
Although the mutations identified in biallelic BRCA2 cases appear to be similar in type and position to those in heterozygous cases, it is noteworthy that three of 17 families and five of 23 cases carry BRCA2 886delGT, and all developed brain tumours. Indeed four of six medulloblastomas identified in biallelic BRCA2 cases carry the 886delGT mutation. This mutation has been reported in multiple familial breast cancer pedigrees, but there are several BRCA2 mutations that are known to occur at similar or greater frequency which have not been identified in biallelic BRCA2 cases. It is difficult to conceive a biological explanation for why a particular truncating mutation should be associated with a specific cancer risk. However, scrutiny of the cancer phenotypes and mutations in additional biallelic cases will be of interest to further evaluate this observation. Interestingly, no case homozygous for BRCA2 6174delT has been reported, despite the prevalence of this mutation in ∼1% of the Ashkenazim.22 This may be due to chance, but it is also possible that the phenotype associated with biallelic 6174delT mutations is either more or less severe than for other combinations of mutations such that they have thus far not been identified. It is also notable that BRCA2 heterozygotes do not appear to be at increased risk of childhood cancer. In 173 BRCA2 families ascertained by the Breast Cancer Linkage Consortium there were no cases of WT, brain tumours, or childhood leukaemia in confirmed mutation carriers (D Thompson and DF Easton, personal communication on behalf of the Breast Cancer Linkage Consortium).
None of the reported families with biallelic BRCA2 cases had been investigated for BRCA2 mutations because of a family history of breast cancer prior to the birth of an affected child. In part this may be because affected children present before their parents, as exemplified by WILMS2, in which the mother and paternal aunt developed breast cancer after the boys had died. Thus, although a family history may be helpful in the identification of some biallelic BRCA2 families, it will likely need to extend to at least three generations to detect the cancer history, particularly on the paternal side. The lack of clear diagnostic features of FA, an atypical cancer spectrum, and the absence of a strong family history of cancer suggest that a high index of suspicion may be required to identify children with biallelic BRCA2 mutations.
Acknowledgments
We thank the members of WILMS2 for participating in the research and the many members of the Familial Wilms Tumour Collaboration and the Familial Breast Cancer Susceptibility Collaboration (UK) who have provided samples for this research. We thank David Betts for cytogenetic analyses in WILMS2 and Douglas F Easton for helpful discussions.
REFERENCES
Footnotes
-
This research was funded by the Institute of Cancer Research (UK) and by Cancer Research UK.
-
Conflict of interest: none declared.
-
The Familial Wilms Tumour Collaboration includes the following members: Laura Arbour, Catherine Bonaïti-Pellié, Lisa Cannon-Albright, Agnes Chompret, Trevor Cole, Catharina Dhooge, W Dupuis, Annabel Foot, William Foulkes, Hazel Galvin, Mary Gerrard, Astrid Gnekow, Norbert Graf, Derek King, Judith Kingston, Jan Kohler, Gill Levitt, Ian Lewis, Anne O’Meara, F Millot, Barry L Pizer, Helen Price, Pascal Pujol, Brigitte Royer-Pokora, Valerie Schumacher, Charles Schwartz, Rosemary Shannon, Eamonn Sheridan, Jane Skeen, Patricia Tonin, Gordon Vujanic, Angela Weirich, and Denise Williams.