Article Text

Statistics from Altmetric.com
- ARMS-PCR, amplification refractory mutation system PCR
- DAPI, 4,6-diamidino-2-phenylindole
- DHPLC, denaturing high-performance liquid chromatography
- FISH, fluorescence in situ hybridisation
- FITC, fluorescein isothiocyanate
- PCR, polymerase chain reaction
CHARGE association (MIM 214800) is a sporadic disorder, characterised by coloboma of the eye, choanal atresia, cranial nerve dysfunction, characteristic external and inner ear abnormalities, cardiac anomalies, genitourinary abnormalities, and growth retardation. Various chromosomal rearrangements have been reported in rare patients with a CHARGE-like phenotype. These include balanced translocation between chromosomes 6 and 8,1 unbalanced translocations involving chromosomes 2 and 18, 3 and 22,2 partial trisomy of 19q with partial monosomy 21q,3 inverted duplication of chromosome 14 (14q22→q24.3),4 and partial trisomy of 2q.5 Based in part on the inconsistent chromosomal aberrations in rare patients with CHARGE association, it is most likely that this condition is genetically heterogeneous. Within the group of children with CHARGE association, there is clearly a subgroup with distinctive clinical characteristics that appears to have a recognisable syndrome.6 Previously, a systematic scan for loss of heterozygosity using microsatellite markers in 10 such patients failed to identify a discernible submicroscopic deletion.7 Although several candidate genes such as PITX28 and PAX29 have been investigated, no mutations have been identified in patients with CHARGE syndrome. Here, we demonstrate a de novo mutation in SEMA3E in an affected patient, identified upon mapping the translocation breakpoints in an unrelated individual with a de novo balanced translocation involving chromosomes 2 and 7: karyotype 46,XY,t(2;7)(p14;q21.11).
METHODS
The study sample includes 72 patients, of whom 43 have either four major criteria for CHARGE (coloboma, choanal atresia, characteristic ear abnormality, and cranial nerve dysfunction) or three major and three minor criteria (genital hypoplasia, developmental delay, cardiovascular malformations, growth deficiency, orofacial cleft, tracheo-oesopageal fistula, and characteristic face) as described by Blake et al.10 This set of patients was used for all FISH analyses, DHPLC, and candidate gene sequencing. An additional 29 patients with clinical diagnosis of CHARGE, established by one of several experienced dysmorphologists,7 were also included in this study. The spectrum of defects in all these patients has been reported previously.7 The phenotype of the child with the balanced translocation has been described by Martin et al11 and includes bilateral choanal atresia, absence of semicircular canals, cranial nerve dysfunction, genital hypoplasia, developmental delay, and growth retardation. A blood sample was obtained from this patient and lymphoblastoid cell line was established. The research protocol was reviewed and approved by the Baylor College of Medicine Institutional Review Board.
To map the breakpoints, we performed fluorescence in situ hybridisation (FISH) on metaphase chromosomes. BAC and PAC clones were obtained from the Children’s Hospital Oakland Research Institute BACPAC Resources Center. CTD-2200N21 was requested from Research Genetics. FISH analysis was carried out as previously described.7 After hybridisation and washing, biotinylated probes were detected by avidin-FITC and digoxigenin probes were detected using antidigoxigenin antibody coupled to rhodamine. The chromosomes were counterstained with DAPI and analysed with a Zeiss Axioskop fluorescence microscope. Long range PCR product was generated using an Expand Long Template PCR Kit (Roche). SEMA3E and SEMA3C were directly sequenced using a BigDye Terminator Sequencing Kit (Applied Biosystems). DHPLC followed by sequencing was used to screen for mutations in PCLO.
Key points
-
CHARGE syndrome is a readily recognisable condition, characterised by choanal atresia, coloboma of the eye, cranial nerve dysfunction, characteristic external and inner ear abnormalities, cardiac defects, genitourinary abnormalities, and growth retardation.
-
Although much evidence points towards genetic causation, the aetiology of this condition remains unknown.
-
We mapped the translocation breakpoint in a patient with CHARGE syndrome, with a de novo balanced translocation involving chromosomes 2 and 7, and identified SEMA3E within 200 kb of the translocation breakpoint on 7q21.11.
-
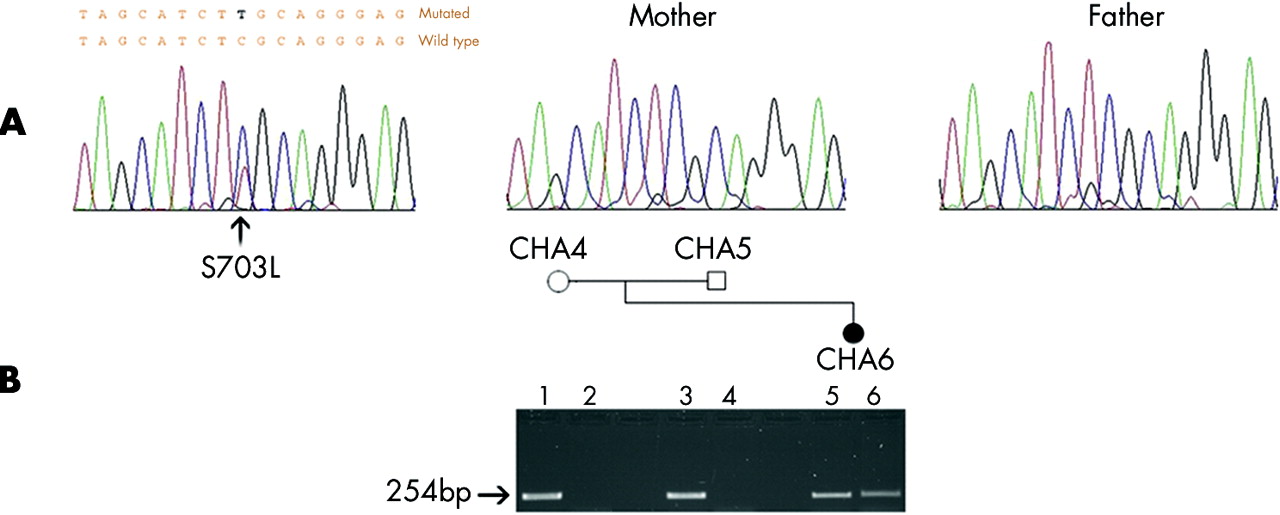
Sequencing additional patients for mutations in SEMA3E revealed a de novo mutation in an unrelated patient, with substitution of the conserved serine residue at 703 to leucine (S703L). This was not detected in either the parents or the 338 ethnically matched control chromosomes.
-
We propose that in a subset of patients with CHARGE syndrome, a common molecular mechanism involving SEMA3E may play an important role in the pathogenesis of the disease.
RESULTS
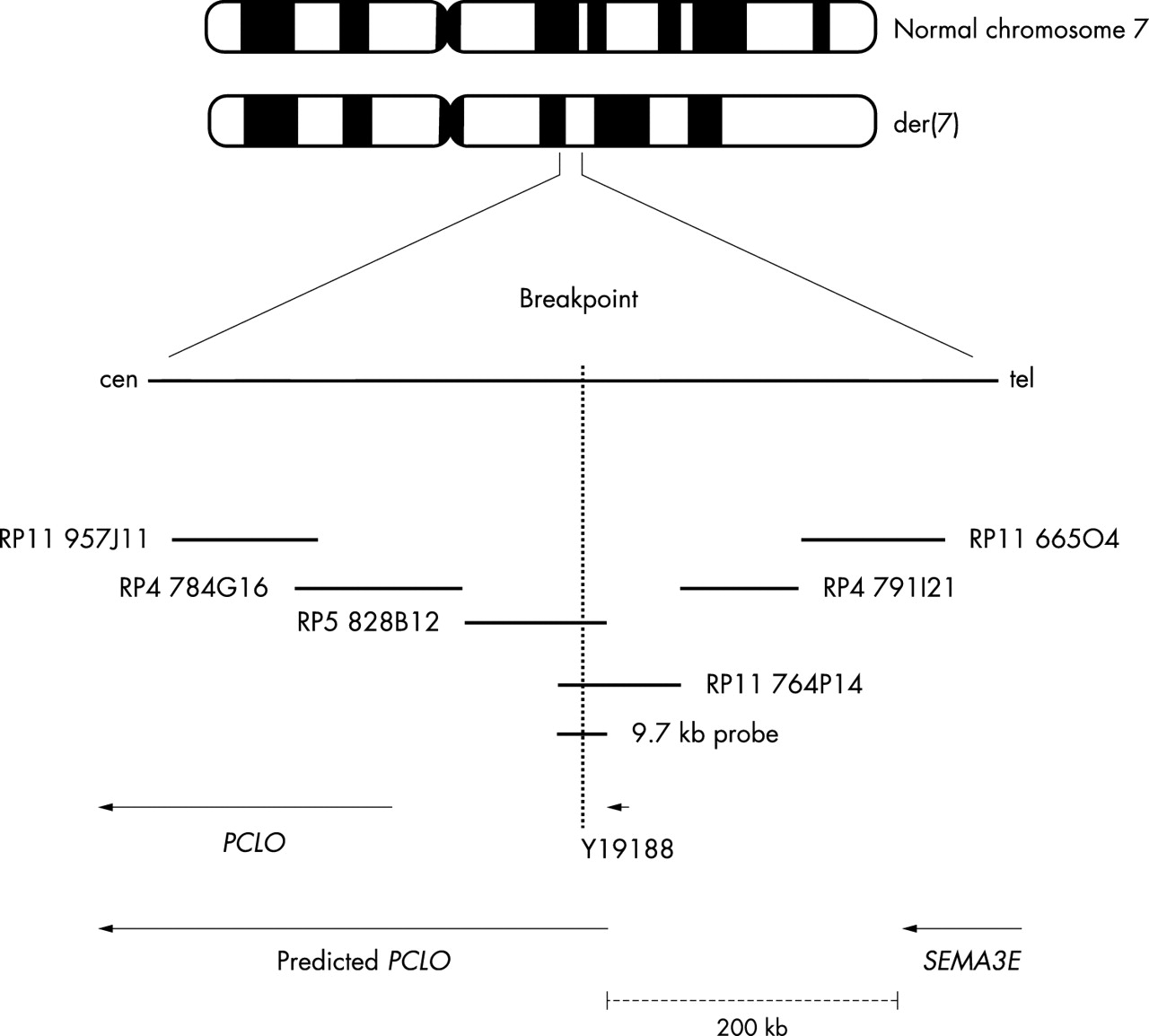
PAC clone RP5 828B12 (GenBank accession no. AC004903) and BAC clone RP11 764P14 (GenBank accession no. AC093461) were both found to span the breakpoint on chromosome 7q21.11 (fig 1). BAC clone CTD-2200N21 (GenBank accession no. AC092571) spans the interrupted segment on chromosome 2p14. The breakpoint on chromosome 2 does not appear to disrupt a gene. However, using a long range PCR fragment of 9.7 kb derived from RP5 828B12, the breakpoint on 7q21.11 was localised to the intronic sequence of the predicted PCLO gene between exons 3 and 4 (fig 2). Piccolo is a large scaffolding protein in the presynaptic active zone that shares homology with Rim and Bassoon.12 It is known to be involved in the trafficking of neurotransmitters at the synapses. We screened 24 patients for mutations in PCLO using DHPLC followed by DNA sequencing, and found no PCLO mutations. To exclude a submicroscopic deletion at 7q21.11, we used RP11 764P14 for FISH to hybridise 30 CHARGE patients. None of these 30 patients was found to have a discernible deletion within this interval. SEMA3E is approximately 200 kb telomeric to PCLO. Sequencing of 72 patients for the SEMA3E gene revealed a missense mutation with a C→T change at bp 2108 in one patient. This causes serine to leucine substitution at amino acid residue 703 (S703L) (fig 3). This change was not seen in either parent of the proband, nor in 338 ethnically matched control chromosomes. Another patient was found to have a non-synonymous change, C1855T, resulting in the missense mutation Arg619Cys. This sequence change was also present in an unaffected parent, but not present in 164 control chromosomes. The non-synonymous SNP Arg208Pro in SEMA3E was seen in both cases and controls with allele frequency of 0.11 and 0.15, respectively. There was no over-transmission of this variant. The coding sequence variant I717V was detected in one African-American patient, but was found to have an allele frequency of 0.095 in ethnically matched control samples. Table 1 summarises the polymorphisms identified in the coding region of SEMA3E. RP11 665O4 and RP5 1102B4 clones containing the DNA sequence of SEMA3E were also used for FISH analysis in 30 patients. None of these patients showed a detectable deletion of SEMA3E using these clones.
Summary of polymorphisms in SEMA3E
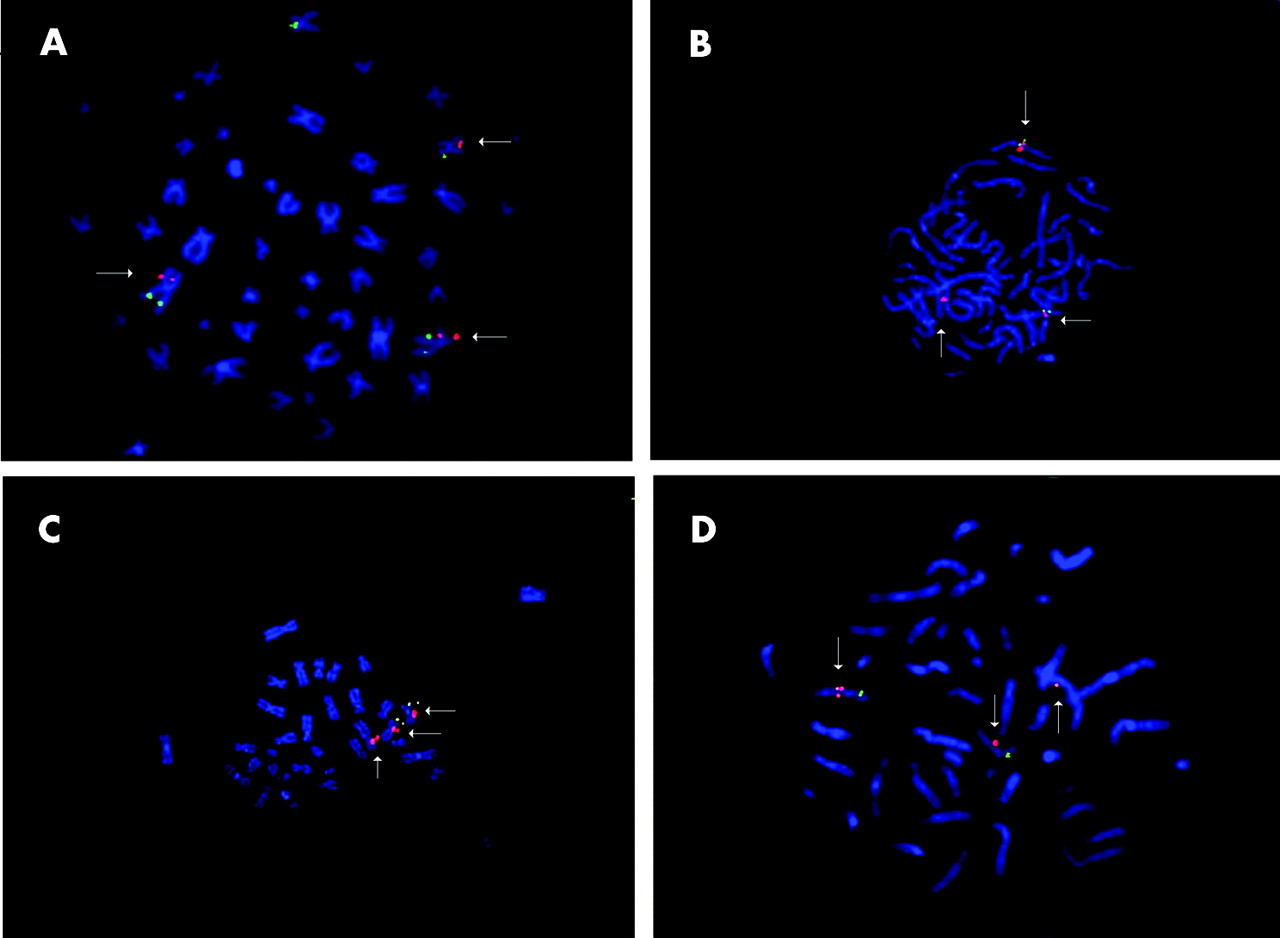
Fluorescence in situ hybridisation showing (A) CTD-2200N21 spanning the translocation breakpoint on chromosome 2. (B) and (D) show the spanning BAC clone RP11 764P14 and PAC clone RP5 828B12 on the chromosome 7 breakpoint (arrows indicating three red signals). (C) Figure demonstrates three signals with 9.7 kb long range PCR probe, amplified from RP5 828B12 on chromosome 7.
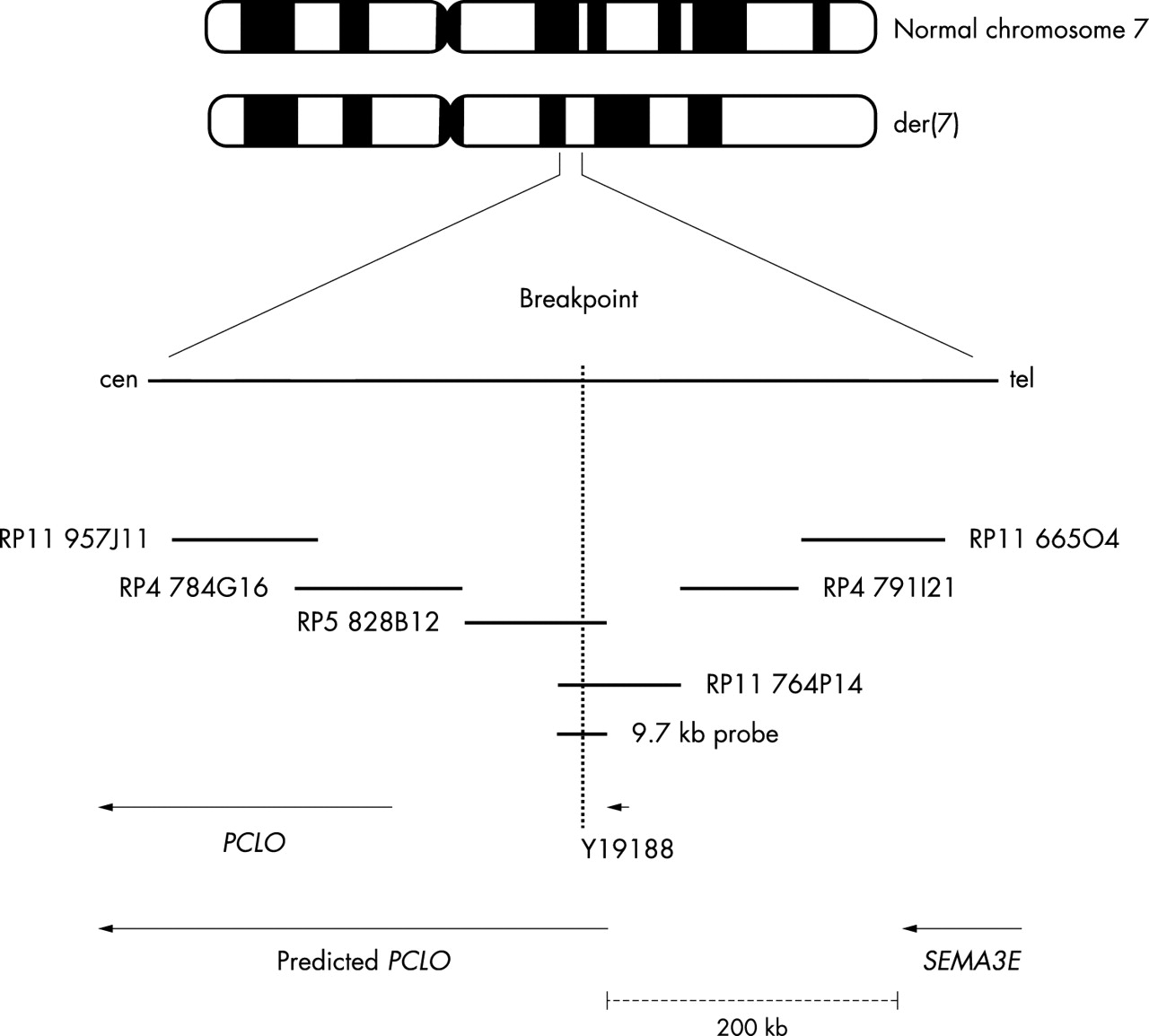
Map of the translocation breakpoint on chromosome 7.
{kind=link}
{kind=link}
{kind=link}
(A) De novo missense mutation resulting in S703L in the affected patient. (B) The gel below the pedigree of individual CHA6 shows amplification of wild type allele (lanes 1, 3, 5) by amplification refractory mutation system PCR (ARMS-PCR). Amplification of the mutant band is observed in the patient (lane 6), but not seen in the parents (lanes 2 and 4).
DISCUSSION
Here we demonstrate a de novo chromosomal translocation adjacent to SEMAPHORIN 3E in one patient, and a de novo missense mutation of a conserved amino acid in the same gene in an unrelated child. This is the first description of a mutation in a patient with CHARGE syndrome. The phenotype of the child with the de novo missense mutation includes iris coloboma, choanal atresia, ear malformation, profound deafness, tetralogy of Fallot, developmental delay, and growth retardation.
Semaphorin proteins are involved in a variety of cellular processes, including axon guidance and cell migration. Semaphorin signalling plays a critical role in neural crest mediated heart development through its interaction with PlexinA2.13Sema3C null mice die from persistent truncus arteriosus and interruption of the aortic arch secondary to abnormal migration of cardiac neural crest cells.14SEMA3C lies 1.9 Mb centromeric to the PCLO gene and, based on the functional data, was also sequenced in 24 CHARGE patients. Various synonymous changes were seen in SEMA3C including T1287C, G1524A, and A2028G. However, no mutations were identified in the coding region in this subset of patients. It has been postulated that the anomalies seen in CHARGE result from abnormal development, migration, or interaction of neural crest cells in early embryogenesis.15 Based on the involvement of otic and nasal olfactory placodes and cranial nerves, another hypothesised mechanism in CHARGE is aberration of placode induction. Halloran et al16 have shown expression of semaZ2 in several zebra fish placodal structures, including nasal placode, otic placode, and the migrating primordium of the lateral line. Inner ear abnormalities including the absence of semicircular canals are now a well-recognised feature in CHARGE.17 Miyazaki et al18 have shown that M-SemaH, the mouse homolog of SEMA3E, is highly expressed in the epithelium of semicircular duct walls during embryonic stages. Based on these data, we believe that SEMA3E may play an important role in the pathogenesis of this syndrome. There are various reasons to regard this mutation in our patient as pathogenic. First, it is a de novo mutation. Second, this change has not been identified in 338 unrelated control chromosomes. Third, this amino acid is conserved in human, mouse, and rat (fig 4). The non-synonymous change C1855T, resulting in missense substitution of the conserved amino acid residue arginine to cysteine (R619C) in another patient and his unaffected parent may reflect a very rare polymorphism, however, the possibility of incomplete penetrance in this family cannot be disregarded. The phenotype of this patient includes retinal coloboma, choanal atresia, ear malformation, hearing loss, facial palsy, atrial septal defect, and growth retardation. The considerable phenotypic heterogeneity observed in patients with CHARGE could reflect separate underlying molecular mechanisms. However, in a subset of these patients, aberration of a common pathway is proposed with SEMA3E as one protein member in this cascade.
{kind=link}
Conserved serine residue of SEMA3E at position 703.
Acknowledgments
We are grateful to the families for participating in the study and thank Margaret A. Hefner for her assistance in patient enrolment. We are also greatly appreciative of the CHARGE Syndrome Foundation for its continued support. We thank Pawel Stankiewicz and Svetlana Yatsenko for assisting in FISH analysis.
REFERENCES
Footnotes
-
Support for this work was provided by the Doris Duke Clinical Scientist Development Award and NIH Award HD39056.
-
Conflict of interest: none declared.