Article Text

Statistics from Altmetric.com
Long QT syndrome (LQTS) is a prototypic arrhythmic disorder that is characterised by prolonged QT interval (or QTc) on electrocardiograms (ECGs), syncope, and sudden death from episodic ventricular tachyarrhythmias, specifically torsade de pointes.1–4 LQTS causes sudden deaths in young, otherwise healthy, individuals, and in many cases the first symptom is sudden death. Both genetic and acquired factors contribute to the pathogenesis of LQTS. Predisposing genetic mutations have been identified in six genes. These include the cardiac potassium channel genes KvLQT1 or KCNQ1 (chromosome 11p15.5, LQT1),5–7HERG or KCNH2 (7q35–36, LQT2),8KCNE1 (21q22, LQT5),9,10 and KCNE2 (21q22, LQT6),11 the cardiac sodium channel gene SCN5A (3p21–24, LQT3),12,13 and the non-ion channel ankyrin-B gene encoding a protein that links ion channels to the cytoskeleton (4q25–27, LQT4).14
Acquired long QT syndrome (aLQTS) is LQTS caused by factors such as bradycardia, cardiac ischaemia, metabolic abnormalities (including hypocalaemia and hypomagnesaemia), starvation (anorexia nervosa), and various medical manipulations and medications including general anaesthetics, antibiotics, antihistamines, and ironically anti-arrhythmic agents.15,16 Acquired LQTS is common, with a population prevalence rate of up to 8%.17 Because almost all cases of acquired LQTS are sporadic, genetic analysis of acquired LQTS has been lagging behind inherited LQTS. However, there has recently been increased interest in determining the genetic basis of acquired LQTS by studying genes causing inherited LQTS.18–21 We carried out a similar analysis in this study.
Voltage gated sodium channels are transmembrane proteins responsible for generating cardiac action potentials, and for rapid conduction of electrical signals through cardiac tissues. The cardiac sodium channel is a large protein of 2016 amino acids encoded by the SCN5A gene.22 The cardiac sodium channel consists of a pore forming α-subunit composed of four homologous domains (I–IV), each containing six transmembrane segments (S1–S6).22 SCN5A is central to the genesis of cardiac arrhythmias and sudden death, and its mutations cause inherited LQTS,13 idiopathic ventricular fibrillation and Brugada syndrome,23 and cardiac conduction disease.24 In this report, we have identified an SCN5A mutation, R1193Q (R/Q), in one of seven patients with acquired LQTS. Functional studies demonstrate that the electrophysiological severity of R/Q is nearly identical to two LQT3 mutations, N1325S (N/S) and R1644H (R/H), which cause susceptibility to inherited LQTS.
MATERIALS AND METHODS
Human genomic DNA samples
Informed consent was obtained from study participants in accordance with standards established by local institutional review boards. Genomic DNA was prepared from whole blood with the DNA Isolation kit for Mammalian Blood (Roche Diagnostic Co., Indianapolis, IN, USA).
Key points
-
Long QT syndrome (LQTS) is a cardiac disorder that causes syncope and sudden death from ventricular tachycardia torsade de pointes. Mutations in the cardiac sodium channel gene SCN5A cause the inherited type 3 LQTS (LQT3). LQTS can also be caused by drugs and other factors, known as acquired LQTS.
-
In this study, we identified a missense mutation, R1193Q or 3578G→A in SCN5A in one of seven patients with acquired LQTS.
-
Mutant R1193Q SCN5A channels destabilise inactivation gating and generate a persistent, non-inactivating current that is expected to prolong the cardiac action potential duration, leading to LQTS. Single channel recording revealed that the molecular mechanism for the generation of the late persistent inward current is frequent, dispersed reopening of the channels.
-
The biophysical defects of R1193Q are nearly identical to two well characterised SCN5A mutations, N1325S and R1644H, which cause LQT3.
-
These results strongly suggest that R1193Q is a functional mutation that can increase susceptibility to LQTS.
-
Notably, 0.2% of the general population carry the SCN5A mutation R1193Q, suggesting that this mutation may be an important risk factor to LQTS in the general population.
The control population consists of 90.5% whites, 3.7% African Americans, 0.8% Hispanics, 0.4% Asians, 0.4% Native Americans, and 4.2% others.
Single strand conformation polymorphism (SSCP) and DNA sequencing analyses
PCR primers to amplify exons and intron−exon boundaries of SCN5A were as described previously.25 SSCP and DNA sequencing analyses were also carried out as described previously.5,12,23,26
Mutagenesis
The mutation (R/Q) was introduced into the wild type SCN5A expression construct by site directed mutagenesis using the mega-primer PCR based method,27 and verified by DNA sequencing.
Electrophysiology and data analysis
Electrophysiological recording was performed in human embryonic kidney (HEK-293) cells transiently transfected with hH1 cDNA (no H558R or Q1077del variants) as described previously28,29 and in Xenopus oocytes injected with in vitro transcribed cRNA as described previously.30
RESULTS
Identification of SCN5A mutation R/Q in a patient with acquired LQTS
The complete genomic structure of SCN5A has been determined previously,25 and intronic primers are available for amplifying all exons and exon−intron boundaries by PCR. Using SSCP analysis, we identified an anomalous band in patient P20680 who was affected with acquired LQTS (fig 1A). DNA sequence analysis of the normal and the aberrant SSCP conformers revealed that the abnormal SSCP conformer contained a single base substitution (G to A transition; 3578G→A) at the second nucleotide of codon R1193 of SCN5A (fig 1B), which resulted in a non-conservative replacement of arginine with glutamine (R/Q) between transmembrane domains II and III of the cardiac sodium channel (fig 1C). We also screened patient P20680 for mutations in other known LQTS genes, but found none (data not shown). As the index patient P20680 is an 82 year old single male without living family members, we were unable to determine whether other family members carry the R/Q mutation.

SCN5A R/Q missense mutation in a patient with acquired LQTS and ventricular tachyarrhythmias. (A) The results of SSCP analyses. The aberrant SSCP conformers are indicated by arrows. Sequence analyses of the normal (left) and aberrant conformers (right) revealed a G→A substitution (B), which results in substitution of arginine at codon 1193 by glutamine in the linker between homologous domains II and III (C). (D) Polymorphic ventricular tachycardia recorded in patient P20680 by Holter monitoring. Atrial fibrilliation (AF) was also detected in patient P20680, but it remains to be determined whether SCN5A mutation R1193Q is the cause of AF in this patient.
The index patient P20680 is a white male, and developed LQTS (QTc=490−570 ms) with administration of d-sotalol, whereas the baseline QTc before drug treatment was 442 ms. Two days after d-sotalol was stopped, a Holter report detected monomorphic ventricular tachycardia (VT) (fig 1D). No polymorphic VT or torsade de pointes was detected. The patient was then started on quinidine; this treatment also produced dramatic QT prolongation (QTc=508 ms).
High population prevalence rate of SCN5A R/Q
To test whether SCN5A R/Q is present in the general population, SSCP analysis with the primers used to identify the R/Q mutation was performed using DNA samples from 2087 randomly selected individuals, a predominantly white population. Anomalous SSCP conformers were identified in 4 of 2087 individuals (0.2%). We sequenced these abnormal conformers and found that they contained the R/Q mutation. These data suggest that the average prevalence rate of the R/Q mutation in the general population can reach up to 0.2%.
Among the four carriers with SCN5A R/Q from the general population, we were able to obtain ECGs from only two, who had a QTc of 421 ms and 430 ms, respectively. It would be useful to determine whether the QTc of these carriers with SCN5A R/Q would become more prolonged by quinidine and other drugs, but such studies raise ethical issues.
Establishment of R/Q as a functional mutation
The R/Q mutation results in the neutralisation of a highly conserved, positively charged side chain within the intracellular domain of the channel protein. Previous studies have shown that similar substitutions are responsible for altered channel gating.30,31 To determine whether SCN5A R/Q is a functional mutation, we used electrophysiology to characterise it by heterologous expression in Xenopus oocytes and in mammalian HEK293 cells.
Fig 2A, B shows representative intracellular microelectrode voltage clamp records for either wild type (A) or R/Q (B) channels. Current–voltage families were evoked by test pulses of −50 to +40 mV (10 mV increments) from a holding potential of −100 mV. A comparison of the peak current−voltage relationships in the two channels (fig 2C) showed that the voltage dependence of activation was nearly unchanged by the R/Q mutation (open squares). Both sets of data accurately fitted to Boltzmann expressions (smooth curves) with a midpoint potential of −29 mV and slope factor of 5.7 mV. The most striking feature of R/Q channels was that Na+ currents at moderately depolarising test potentials (−40 to 0 mV) decayed more rapidly than the normal time course (fig 2A, B). As the onset of inactivation is the major contributor to the decay of Na+ current at test potentials more positive than −40 mV,32 we fitted the time course to a mono-exponential decay function and plotted the time constant of inactivation (τinact, log scale) as a function of test potential (fig 2D). At negative test potentials, the onset of inactivation was significantly faster in R/Q (open squares) than wild type (filled circles) channels (for instance, a threefold acceleration of R/Q time constants was obtained at a test potential of −40 mV). The voltage dependence of steady state inactivation (fig 2E) was consistent with the observed kinetic changes; compared with wild type channels, R/Q channels displayed a negative shift of the steady state inactivation curve. When fitted to Boltzmann functions (smooth curves) a midpoint potential of −70 mV (slope factor 5.5 mV) was obtained in wild type, and −76 mV (slope factor 5.3 mV) in R/Q (fig 2E).

Electrophysiological characterisation of normal (A) and R/Q mutant (B) Na+ channels. Whole cell currents (A–B) were obtained by intracellular microelectrode voltage clamp. Each panel shows superimposed current traces evoked by test pulses of −40 to 0 mV (10 mV increments) from a holding potential of −100 mV. Linear leakage and capacitation were corrected by online (P/4) subtraction; residual transients were blanked. Peak test pulse current amplitude (normalised to maximum current, mean (SEM)) was plotted as a function of test pulse amplitude for wild type (WT; filled circles) and R/Q (open squares) channels. The data were fitted to Boltzmann distributions of the form: I = Gmax(V−Vrev)(1−(1/(1+exp((V−V0.5)/k)))); where Gmax = scale factor, V = test pulse amplitude, Vrev = reversal potential, V0.5 = midpoint potential, and k = slope factor. The decay phase of current evoked by test pulses of −40 to +40 mV was fitted to mono-exponential decay functions and the time constant (τinact) was plotted semi-logarithmically v test pulse potential (D). The voltage dependence of steady state inactivation was determined using a standard two pulse protocol in which a 500 ms variable amplitude conditioning pulse was followed by a 15 ms fixed amplitude (−10 mV) test potential. Test pulse current amplitude (normalised to maximum current) was plotted v conditioning pulse amplitude to assess the fraction of non-inactivated channels (E). The data were fitted to Boltzmann distributions (smooth curves) of the form: I = (1+exp((V−V0.5)/k))−1 where V = conditioning pulse amplitude, V0.5 = midpoint potential, and k = slope factor. Pooled data (C−E) are plotted as mean (SEM), n = 8, and 18, respectively for WT and R/Q.
Mutations that produce persistent non-inactivating Na+ currents cause LQT3.31 We tested for the presence of such currents at the whole cell level by a tetrodotoxin (TTX) subtraction method.33 Examination of the late phase (time >30 ms) of current at high amplification (peak current off the scale) revealed negligible inward Na+ current in wild type channels (A) but substantial late, non-inactivating current in R/Q channels. The increase in persistent current in mutant channels was not due simply to an over-expression of R/Q channels, as peak WT (fig 3C) current was larger than that of R/Q (fig 3D), presumably because of variations in expression level. We compared the relative amounts of persistent current by normalising to peak current in each cell (fig 3E). Wild type cells produced negligible levels of persistent current, but R/Q mutants produced substantial amounts of persistent current that were comparable to that produced by other LQT3 mutants, R/H and N/S, and about half of that produced by the most severe LQT3 mutant, ΔKPQ.30 Similar results were obtained in a mammalian expression system (transiently transfected HEK-293 cells). Test pulses of 300 ms duration evoked a persistent inward current (mean (SEM), n = 10 cells) of −44.3 (7.2) pA in cells that expressed R/Q. In contrast, the amplitude of persistent currents in cells that expressed wild type channels was −17.4 (10.0) pA (n = 12). When normalised to peak current amplitudes, the level of persistent current in R/Q mutant channels was 1.29 (0.33)% of the peak amplitude v 0.31 (0.30)% in wild type channels. These results suggest that the SCN5A R/Q is a functional mutation, and that, similar to the well characterised R/H and N/S LQTS mutations, R/Q is expected to increase the risk of LQTS.

Late Na+ current in normal and mutant channels. Records of inward Na+ current (at a −10 mV test potential) were obtained by subtracting records obtained after application of 200 mmol/l TTX from total current before TTX. Typical difference currents obtained in this way are illustrated for WT (A and C) and R/Q mutant (B and D) channels. High amplification (A and B) revealed significant amounts of persistent late current that represents a small fraction (<1%) of the peak current (C and D, note the change in amplification and time base) only in the mutant channels (B). Panel C presents the pooled data of each channel group (WT and R/Q), compared with ΔKPQ.30 Persistent current (normalised to peak currents and expressed as mean (SEM) per cent) was taken as the average current over the time interval 180–200 ms (except for ΔKPQ, measured at 300 ms). The number in parentheses is the number of oocytes tested. *Indicates significant difference between means (p<0.05, two tailed Student’s t test) compared with WT.
We have shown previously that dispersed reopenings are responsible for nearly all of the persistent current in the LQT3 mutants R/H and N/S, and about half of that in ΔKPQ.30 The R/Q mutant associated with acquired LQTS showed a pattern similar to the R/H and N/S mutants of inherited LQTS (fig 4). Thus, in multi-channel, cell attached patches during maximum activation, wild type channels rarely reopened after the initial transient current (off scale) of synchronously active channels had subsided. An example of a rare reopening is shown in trace A1. By contrast, R/Q channels (fig 4B1–B4) generated frequent, dispersed reopenings similar to those observed previously in the LQT3 mutants. In R/Q channels, as in wild type (and in the previously described R/H and N/S mutants), prolonged bursting such as that associated with the ΔKPQ mutation was extremely rare.

Single channel basis of persistent current in R/Q mutant channels. Current records were obtained using extracellular patch clamp recording in the cell-attached mode. (A, C, E) Wild type (WT); (B, D, F) R/Q. Consecutive current traces (A1–4; B1–4) were evoked by a test potential of 0 mV from a holding potential of −100 mV (preceded by a 100 ms prepulse to −140 mV to remove resting inactivation) at a frequency of 1 pulse/s. Channel openings are downward deflections in the current traces. The start of the test pulse is coincident with the residual uncorrected capacitative current. Panels C and D show ensemble averages of 128 records, plotted at a high amplification to emphasise the persistent current observed in R/Q channels (D). Peak currents were off the scale. Panels E and F show fast time base, low amplification plots of the ensemble averages to facilitate comparison of peak currents. Recording bandwidth was 2 kHz. Similar results were obtained in replicate experiments from 11 WT and 7 R/Q patches.
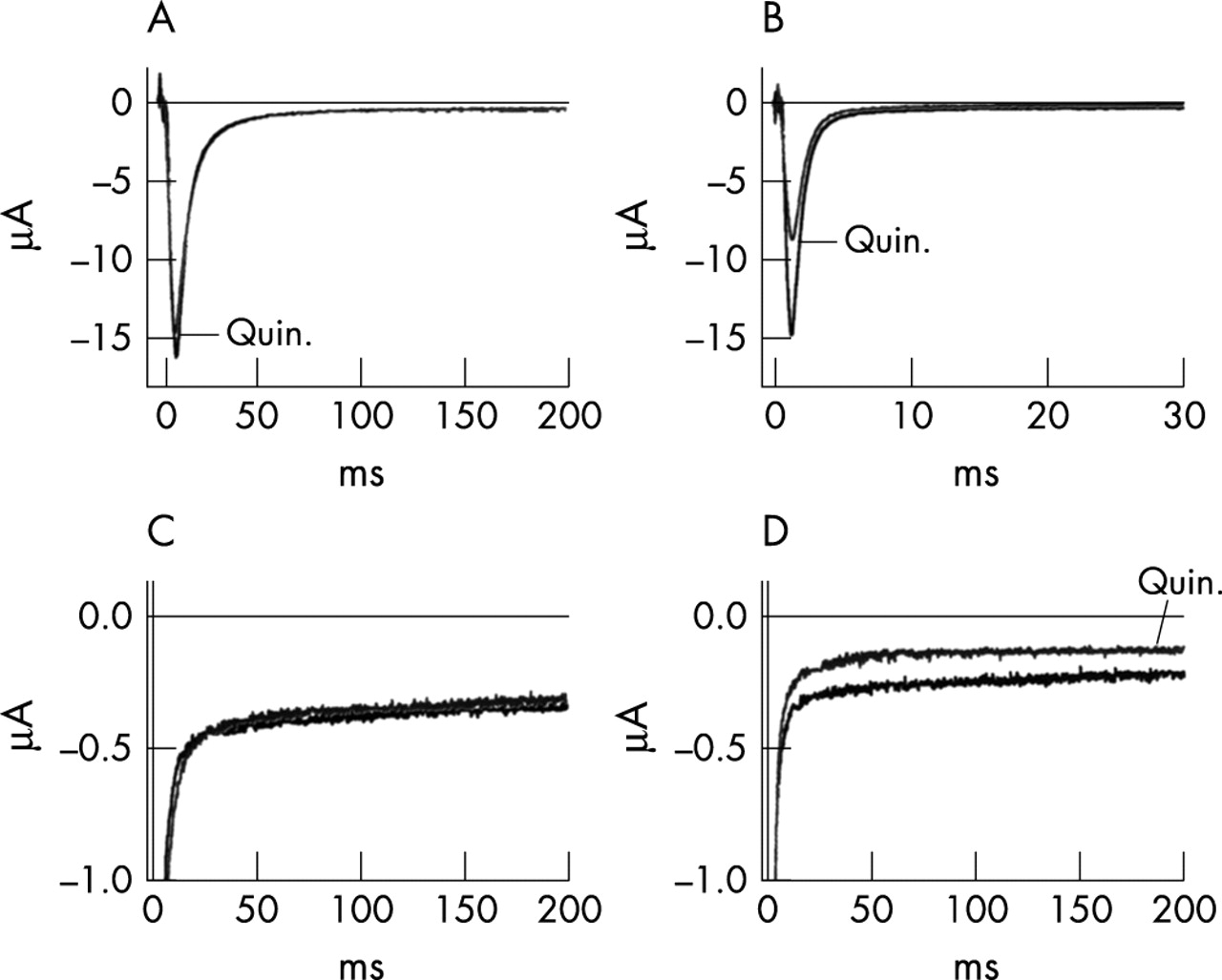
Because acquired LQTS in this patient was precipitated by quinidine, a drug that blocks both Na+ and K+ channels, we were interested to know whether the late currents generated by R/Q mutant channels were affected by quinidine treatment. Quinidine blocks Na+ channels in a use dependent manner such that repetitive brief depolarisations that maximally activate channels produce the greatest blocking effect.34 As shown in fig 5A, quinidine at 50 mol/l had little effect on peak Na+ conductance in resting R/Q mutant channels (that is, in channels maintained for several minutes at the holding potential of −120 mV without repetitive activation). Average blocking effect was 7.1 (1.5)% (n = 7). Similar results were obtained in wild type channels (not illustrated), with an average block of 6.3 (0.6)% (n = 7). Most importantly, however, quinidine was ineffective in blocking the late, residual current (fig 5C) in the absence of a conditioning train even though the test pulse was long enough to produce >90% inactivation of the peak current. Average late current blocking effect under this condition was 7.7 (2.2)% (n = 6). However, application of a conditioning train of pulses that allowed accumulation of drug bound open channels caused a substantial blockade of peak R/Q currents (fig 5B); the test pulse current was preceded by a conditioning train of 200 brief activating pulses of 5 ms duration and −10 mV amplitude, delivered at a repetition rate of 10 Hz). Average blocking effect of peak current was 40.0 (3.0)% (n = 8). Under this stimulus regimen, late current observed during a long depolarising test pulse was suppressed to the same extent as the reduction in peak current; the average blocking effect of late current was 44.1 (11.6)% (n = 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of quinidine on R/Q channels. (A–B) Representative records of peak currents evoked by test potentials of −10 mV from a holding potential of −100 mV in the presence and absence of 50 μmol/l quinidine. (A) The effect of drug on resting channels held at −100 mV for 3 minutes prior to the test pulse. (B) Maximum blocking effect of activated channels. A conditioning train of 200 brief pulses (5 ms duration, −10 mV amplitude, 10 Hz. repetition rate) preceding the test pulse (50 ms interval at −100 mV) allowed use dependent accumulation of channels in the activated, drug bound state.34 (C–D) Late currents evoked by 200 ms test pulses to −100 mV before and after equilibration in 50 μmol/l quinidine in the same experiment as A and B. Records in panel C and D respectively, were obtained in the absence and presence of the conditioning train described above.
DISCUSSION
In this study, we identified a missense mutation R/Q in the cardiac sodium channel gene SCN5A in one of seven patients with acquired LQTS. Electrophysiological characterisation revealed that R/Q altered the function of the cardiac sodium channel (fig 2–5), which clearly demonstrates that SCN5A R/Q is a functional mutation. It is important to note that the biophysical phenotype of the mutant R/Q channels markedly resembles that of well established mutations (N/S and R/H in SCN5A) causing inherited LQTS. All generate a similar number of late inward currents via dispersed reopening, a well established cause of type 3 LQTS.30,31 Expression of the human SCN5A N/S mutant channel in the mouse heart leads to generation of late persistent inward current in myocytes, and development of LQTS, ventricular arrhythmias, and sudden death in transgenic mice.35 Thus, the SCN5A R/Q mutation is expected to cause susceptibility to LQTS. Furthermore, the combination of a genetic mutation and external factors such as d-sotalol and quinidine may unmask the underlying genetic defect, resulting in the development of LQTS and cardiac arrhythmias. This may explain the finding that the QTc for the index patient with the SCN5A R/Q mutation increased dramatically from 442 ms to 570 ms with d-sotalol and to 508 ms with administration of quinidine. An echocardiograph report for the index patient revealed moderate left ventricle enlargement, moderate global hypokinesis, and mild tricuspid and mitral regurgitation, which may also act as external factors that help precipitate the development of acquired LQTS. Our study is consistent with the findings from two other reports. Piippo et al recently showed that QTc for a carrier of the V1667I SCN5A mutation increased from 460 ms to 580 ms with administration of halofantrine.20 An elderly Japanese woman with acquired LQTS and acquired TdP carried an SCN5A mutation, L1825P.36 However, it should be noted that the R/Q mutation, instead of acting as an allelic variant creating a substrate at risk for acquired LQTS, may represent an incompletely penetrant LQTS mutation.
The index patient in this study also carries another SCN5A polymorphism, H558R, which is present in 20–30% of the population; however, lack of family members meant we could not determine whether H558R is in the cis or trans localisation with R1193R. The patient does not harbour the common variant Q1077del.37 The variant H558R has been shown to modulate the effects of other SCN5A mutations including M1766L and T512I.37–39 Future studies may determine whether H558R has any impact on the R1193R mutation.
In a recent publication, Takahata et al.40 described the identification of the R/Q mutation in one of 6 Japanese patients with Brugada syndrome, and one of 48 normal controls. As shown in fig 2, the R/Q channels inactivate more rapidly than wild type (fig 2A,B), and the onset of inactivation was three times faster in R/Q than in wild type channels (fig 2D). These effects are expected to decrease the availability of sodium conductance, which may account for the possibility that R/Q may also be associated with Brugada syndrome. As previously reported, a single mutation, for example, the 1795insD mutation in SCN5A, can be associated with both Brugada and long QT syndromes.41,42
A de novo Na+ channel missense mutation (R1623Q) associated with severe LQTS has been identified.43 This substitution is at an extracellular position in domain IV of the α-subunit. The highly conserved Arg16,23 corresponds in the human skeletal muscle Na+ channel to the site of paramyotonia congenita mutations (R1448H,C).44 In both cardiac45,46 and skeletal muscle47 channels, mutations at this position slow the rate of inactivation and prolong the single channel open time. In contrast, the highly conserved R1193 described here is located on the intracellular side of the channel in the linker segment between homologous transmembrane domains II and III. The R/Q mutation does not slow the rate of inactivation (fig 2), but rather produces a persistent late current that originates as dispersed reopenings (fig 4). Therefore, unlike the extracellular R/Q mutation, which slows the onset of inactivation, the intracellular R/Q mutation appears to reduce the stability of the inactivated state, allowing brief returns from inactivated to open states in a manner similar to that observed in other intracellular LQTS mutations.
R1193 is located in the linker between domains II and III, several residues upstream from the proposed boundary of transmembrane segment DIIIS1. This residue is conserved among mammalian Na+ channel isoforms, but the functional importance of the linker is under active investigation. In a previous study of rat brain Na+ channels, Stühmer et al48 showed that deletion of a large segment of this linker or the introduction of a cut did not cause gross changes in macroscopic gating characteristics. Therefore, this region is unlikely to be directly involved in gating. However, in agreement with our data (fig 2) the voltage dependence of steady state activation was unchanged while inactivation was shifted by about −6 mV. The low amplitude persistent currents that we observed would probably have gone undetected in their experiments because TTX subtraction and single channel recording were not performed. Our results suggest that R1193, together with other residues located at intracellular sites, including LQT3 loci in domains III and IV of the cardiac Na+ channel30 as well as several myotonia loci in domains III and IV of the skeletal muscle Na+ channel,49,50 influence the stability of the inactivated state as evidenced by mutation induced generation of dispersed reopenings. The mechanism whereby amino acid substitutions influence stability is unknown, but for R/Q substitution both a charge neutralisation and a reduction in volume might have an indirect effect on inactivation either by electrostatic or steric mechanisms.
As the patient with acquired LQTS in this study developed LQTS with administration of quinidine, a well known Na+ and K+ channel blocker, we were interested to know whether the late currents generated by R/Q mutant channels were resistant to quinidine blockade. We found that late current could only be blocked after high frequency conditioning stimulation. This result is consistent with the notion that quinidine primarily blocks channels in the open rather than inactivated or resting states of the channel.34 By contrast, the conditions that trigger arrhythmia in patients include bradycardia and long pauses in heart rhythm. Therefore, the ability of quinidine ability to block Na+ channels is unlikely to be responsible for arrhythmias associated with acquired LQTS. A more likely explanation is that quinidine blockade of outward K+ current unmasks the proarrhythmic action of the persistent current in the mutant Na+ channels.
Our results show that quinidine blockade of late, persistent currents was no more effective than blockade of early, transient Na+ currents. Thus, unlike class Ib antiarrhythmic drugs such as mexiletine and lidocaine, which preferentially block inactivated channels51 and selectively suppress late currents in LQT3 mutant channels,30,41,52 quinidine would not be expected to ameliorate LQT3 defects in gating. Whether the phenotype of the R/Q mutation can be corrected by lidocaine or other drugs that stabilise the inactivated state remains to be determined.
In conclusion, we have identified a common mutation R/Q in SCN5A. Our electrophysiological studies establish R/Q as a functional mutation that can cause susceptibility to cardiac arrhythmias. The common occurrence of the R/Q SCN5A mutation underlines the importance of this mutation as a risk factor for LQTS and ventricular arrhythmia in the general population.
Acknowledgments
This work was supported by NIH R01 HL66251 and a grant-in-aid from the American Heart Association Ohio-Affiliate (Q Wang) Bristol-Myers Squibb (M T Keating), and a grant in aid from the American Heart Association with funds contributed in part by the AHA, Northeast Ohio Affiliate (G E Kirsch). We thank J Poruban for proofreading and the members of the Wang Laboratory for help with preparation of control human genomic DNA.
REFERENCES
Footnotes
-
Conflicts of interest: none declared