Article Text

Statistics from Altmetric.com
Split hand/foot malformation type I (SHFM1, OMIM *183600) is an autosomal dominant developmental disorder of limb formation that results in the absence of the central digital rays, deep median clefts, and syndactyly of the remaining digits. Patients with SHFM1 harbour deletions, translocations, and inversions in chromosomal region 7q21–q22.1 The deletions at 7q21–q22 encompass different genomic regions and probably result in a contiguous gene syndrome that includes growth impairment, microcephaly, craniofacial manifestations, hernias, hearing loss, and mental retardation.2,3 Cases with translocations do not show this broad pattern of abnormalities but are associated with hearing loss in most cases.4,5 Split hand/foot malformation type I is the only form of split hand/foot malformation associated with sensorineural hearing loss, and it has been designated SHFM1D (OMIM *605617).5,6 Recently, SHFM1D was shown to result from Mondini dysplasia in a boy with a de novo deletion of about 8.9–17 cM of the paternal chromosome 7q21.1–q21.3.7 So far, microdeletions at 7q21.3 have been described in only two cases: one in a boy with split hand/foot malformation plus mild mental retardation, growth retardation of post-natal onset, and hypotonia and another in a patient with ectrodactyly, ectodermal dysplasia, and cleft lip/palate (EEC) syndrome.8,9 Mapping of the deletion and translocation breakpoints in several patients showed a critical interval of about 1 Mb for the SHFM1 locus at 7q21.3.9 This interval included a 500 kb region that spanned five of seven known translocation breakpoints. In this region, the candidate genes DLX5 and DLX6 (human homologues of the Drosophila distal-less homeobox gene family) and DSS1 (deleted in the split hand/split foot SHFM1 region) were identified. No mutations were detected in patients with sporadic split hand/foot malformation with translocations or two families with split hand/foot malformation, sensorineural deafness, and normal chromosomes who showed linkage to 7q21.6,9,10 Haploinsufficiency through interruption of the gene’s regulatory elements was considered therefore to be the cause of SHFM1. Recently, an animal model of human SHFM1 has been produced in mice by targeted inactivation of Dlx5 and Dlx6 candidate genes.11,12 In this mouse model, the Dlx5/Dlx6–/– genotype resulted in inner ear and severe limb malformations and craniofacial and axial skeletal defects.11
CLINICAL SUMMARY
The index patient was the four year old son of a 28 year old mother and a 32 year old father who were healthy and not consanguineous. The child was born in the 42nd week of pregnancy. His weight at birth was 3630 g, length at birth 50 cm, and fronto-occipital circumference 36.0 cm. The patient was referred to our department at the age of four weeks because of split hand/split foot malformation. He had ectrodactyly of the left hand and both feet, with typical lobster claw (fig 1). The right hand had syndactyly of the third and fourth digits. Further dysmorphic features were dysplastic ears and retrognathia. A hearing test showed profound deafness. Examination by magnetic resonance tomography showed a malformation of the inner ear typical for Mondini dysplasia, including enlargement of the vestibulum and a plump cochlea structure. Cochlea implantation was performed at the left side. To date, psychomotoric development seems normal.
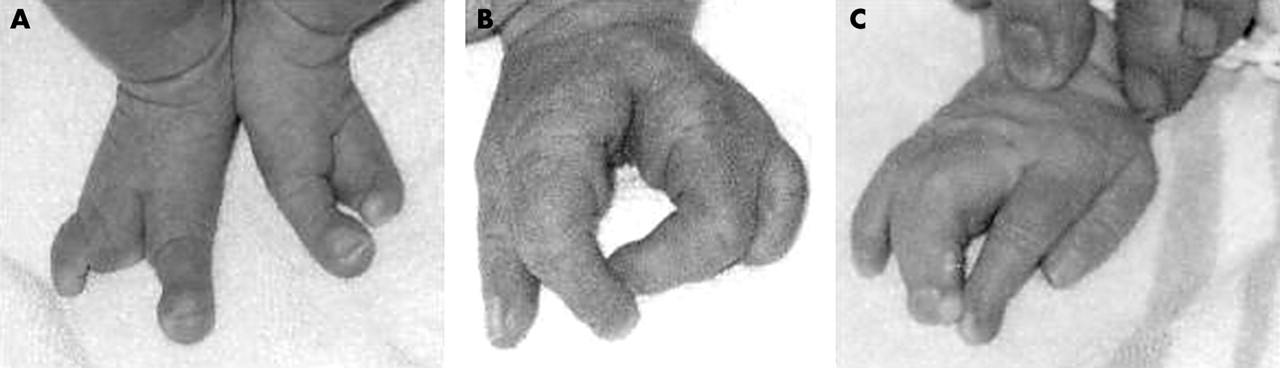
Ectrodactyly of both feet (A) and the left hand (B), with lobster claw malformation. The right hand (C) had syndactyly of the third and fourth fingers.
Key points
-
Split hand/foot malformation type I (SHFM1, OMIM *183600) is characterised by missing digits, syndactyly, and deep median clefts in the hands and feet, and it may be associated with sensorineural hearing loss (SHFM1D, OMIM *605617). The disease locus has been located to chromosome 7q21.3–q22 by the use of deletion mapping and linkage analysis.
-
We report a boy with a de novo microdeletion of 0.9–1.8 Mb in the SHFM1 region, with classical symptoms of SHFM1 and deafness caused by Mondini dysplasia. Haplotype analysis with microsatellite markers showed loss of the paternal alleles D7821, D7S491, and D7624. The microdeletion included the candidate genes DSS1, DLX5, and DLX6, whereas two copies of the flanking DNCI1 gene were retained in the patient’s genome. In all three cases of SHFM1 investigated by haplotype analysis, the paternal alleles were missing.
MATERIALS AND METHODS
Microsatellite analysis
Fourteen microsatellite markers from chromosome 7q21.3–q22.1 retrieved from the NCBI STS map (www.ncbi.nlm.nih.gov/) were informative in the family. Primers for polymerase chain reaction (PCR) were synthesised commercially (MWG-Biotech, Ebersberg, Germany), and standard PCR analysis was performed.13
Southern blot hybridisation
We cleaved high molecular weight DNA with restriction endonuclease Hind III. We resolved the digests on 0.8% agarose gel and blotted them on a nylon membrane (Hybond-N+, Amersham Biosciences, Little Chalfont, UK). We produced hybridisation probes DNCI1 (accession number NM_004411), DLX5 (accession number NM_005221), and DXL6 (accession number NM_005222) by amplification of cDNA templates from placenta and fetal brain tissue (Clontech, Palo Alto, CA, USA) with PCR primers DNCI1for 5′AGAAGAGAAGAAACGGAAGG3′ and DNCI1rev 5′CAGGAACACATTTTGCCATC3′ to give a 1188 bp PCR product, DLX5for 5′CGTCTCAGGAATCGCCAACT3′ and DLX5rev 5′ACTGGTTGGAGGTCGGAGG3′ to give a 651 bp PCR product, and DLX6for 5′ACTCGCAGCACAGCCCTTAC3′ and DLX6rev 5′CTGCATCGTGTCCTGGTGT3′ to give a 513 bp PCR product. Annealing temperatures were 55°C for DNCI1 and 61°C for DLX5 and DLX6. We performed PCR amplifications with the Enhanced High Fidelity PCR System (Roche Diagnostics, Basel, Switzerland). We purified PCR products with a QIAEX II Gel Extraction Kit (Qiagen, Hilden, Germany) and radiolabelled them by random priming with alpha-32P dCTP. We carried out hybridisations, washings, and autoradiography according to standard protocols.14
RESULTS
Chromosome analysis from the phytohemagglutinin stimulated lymphocytes of the patient showed a normal karyotype, apart from a heteromorphism in chromosome 9 (karyotype: 46,XY,9qh–). This heteromorphism was also detected in the mother (46,XX,9qh–). The karyotype of the father was inconspicuous (46,XY). In the patient, no deletion in chromosome 7 could be detected at a banding level of 400, and no abnormal comparative genomic hybridisation pattern was seen (data not shown), which suggests the possibility of a split-hand/foot malformation microdeletion in this patient. Haplotype analysis of the SHFM1 critical region at 7q21.3–q22.1 of the parents and their affected son showed unambiguous loss of the paternal markers D7S821, D7S491, and D7S624, whereas the flanking markers D7S2482 and D7S618 retained heterozygosity (table 1, fig 2). Additional markers within this interval were found to be uninformative. To delineate the deletion further, we performed quantitative Southern blot analysis of the genes DLX5, DLX6, and DNCI1 (dynein cytoplasmic intermediate polypeptide 1) within the interval. For the DNCI1 gene, signal intensity was the same in the affected boy, his parents, and a normal control person (fig 3). In contrast, for the DLX5 and DLX6 genes, signal intensities in the affected boy were reduced clearly to about half those of his parents and the control person (fig 3). These data suggest retention of both parental copies of the DNCI1 gene but loss of one copy of each of the DLX5 and DLX6 genes in the affected boy. From these results, the deletion breakpoints are located between the DNCI1 gene and marker D7S821 at the proximal border and between the DLX5 gene and marker D7S618 at the distal border. This narrows the deletion to 0.9–1.8 Mb (table 1).
Genotype analysis in the SHFM1 interval on 7q21.3–q22.1

Analysis of microsatellite markers from the critical SHFM1 region. Marker positions are listed in table 1. Genotypes of the mother (M), son (S), and father (F) for the indicated markers are shown.

{kind=link}
{kind=link}
{kind=link}
Southern blot analysis of the DNCI1, DLX6, and DLX5 genes with corresponding cDNA probes. Equal amounts of DNA from the son (S), father (F), mother (M), and a control person (1×C) and half the amount (0.5×C) of the control DNA, respectively, were separated by agarose gel electrophoresis. Hind III cleaved phage λ DNA was used to calculate the fragment sizes shown on the left.
DISCUSSION
We report the first patient with a microdeletion in 7q21.3 who shows SHFM1 with Mondini dysplasia. This microdeletion includes the SHFM1 candidate genes DLX5, DLX6, and DSS1. In mice, all three candidate genes apparently are involved in limb formation.9,11,12 The Dlx5 and Dlx6 genes mostly are coexpressed in a unique spatial and temporal pattern. Knockout mice that solely lack the Dlx5 gene show no apparent limb abnormalities.15 Furthermore, the SHFM phenotype was apparent only in Dlx5/6−/− double knockout mice and could be rescued by transgenic overexpression of the Dlx5 gene.11 This suggests that the Dlx5 and Dlx6 genes functionally are redundant in controlling limb and craniofacial development in mice.11,16,17 Interestingly, Dlx5/Dlx6−/− mice have disturbed development of the inner ear. From the mouse model, it seems possible that deletion of DLX5 and DLX6 also causes Mondini dysplasia in SHFM1D. The role of the DSS1 gene in limb formation is less clear. In yeast, deletion of the DSS1 homologue SEM1 induces pseudohypheal growth under differentiating conditions.18SEM1 and DSS1 functionally are conserved, as DSS1 was able to restore the pseudohypheal phenotype in yeast. SEM1 is a vesicle transport protein and seems to act as a negative regulator for exocyst function. The role of membrane traffic in mammalian development, however, is poorly understood.
In contrast to the situation in mice, contiguous loss of one allele of the SHFM1 candidate genes seems to be enough to cause the SHFM1 phenotype in humans. Most deletions in SHFM1 were detected cytogenetically, and only in two previously reported cases was haplotype analysis performed.3,7 Surprisingly, in these two cases, as well as in the case presented here, the de novo deletions occurred on the paternal chromosome 7. This may indicate preferential loss of the paternal alleles in SHFM1. Recently, a new imprinted gene cluster on human chromosome 7q21, which includes the ϵ-sarcoglycan gene and the paternally expressed 10 (PEG 10) gene, has been suggested.19,20 Both genes are located about 2 Mb proximal of the SHFM1 critical region and seem to be imprinted maternally. Linkage analysis in one of two SHFM1D families, however, does not support a role of maternal imprinting in SHFM1 because maternal transmission of the disease was observed for six of nine female family members.6 Haplotype analysis of a larger number of patients with SHFM1 with de novo deletions on chromosome 7 could show whether the paternal chromosome 7 is prone to deletion of the SHFM1 region.
Acknowledgments
We thank Dr G Klingebiel, Berlin, for his expertise on magnetic resonance topography scanning, and G Koch and G Weilepp for technical assistance.
REFERENCES
Footnotes
-
Conflicts of interest: none declared.