Article Text

Statistics from Altmetric.com
- FISH, fluorescence in situ hybridisation
- MR, mental retardation
- XLMR, X-linked mental retardation
- ZNF81, zinc finger 81
X-linked mental retardation (XLMR) has a prevalence of 2.6 cases per 1000 population, accounting for over 10% of all cases of mental retardation. Clinically, XLMR exists in syndromic (MRXS) and non-syndromic (MRX) forms, that is without other distinguishing features.
Non-syndromic X-linked mental retardation (MRX) is a highly heterogeneous group of conditions in which mental retardation (MR) is the only consistent clinical feature in patients. This in contrast to syndromic forms of X-linked mental retardation (MRXS), where MR is associated with recognisable clinical signs such as congenital malformations, neurological features, or metabolic disturbances. Identifying novel genes that are responsible for MRX is difficult due to the heterogeneity of this disorder. At present 30 genes have been identified as playing a role in MRXS. However in MRX, only 15 genes are known to be involved accounting for less than one-fifth of all MRX.1–6 The recent observations that RSK2, MECP2, and ARX play a role in both syndromic and non-syndromic forms of XLMR,7–10 suggest that a molecular basis to strictly separate these two forms is not always present. In addition, careful clinical re-examination of patients with an OPHN1 gene mutation has revealed distinctive phenotypic hallmarks, such as cerebellar hypoplasia, in patients who were previously classified as non-syndromic.11,12
The frequency of causative mutations in any of the 15 MRX genes known today appears to be very low, so in the majority of patients with MRX the genetic cause is still not known. It has been suggested that up to 100 different genes might be involved in MRX.1–4 Seven of the 15 genes have been cloned on the basis of their disruption by chromosomal rearrangements in mentally retarded patients. Recently, it has been predicted that approximately 30% of all mutations underlying MRX are located in the Xp11.2–Xp11.3 interval.1 Only two MRX genes have so far been identified in this region, ZNF415 and PQBP1.6 In this study, we have characterised the breakpoint of a de novo translocation t(X;9)(p11.23;q34.3) in a female patient with severe mental retardation. Since the breakpoint on the X chromosome is within the hotspot interval of MRX genes, it was an excellent starting point for a positional cloning approach to identify a novel candidate gene responsible for MRX. Fine-mapping of the X-chromosomal breakpoint with fluorescence in situ hybridisation (FISH) analysis and sequencing led to the identification of a gene, ZNF81, that was disrupted by the breakpoint. Subsequent mutation analysis in a cohort of MRX families revealed a missense mutation in a family that maps to Xp11.23, supporting the notion that ZNF81 is a novel MRX gene.
METHODS
Propositus
The patient was a Caucasian girl born after 37 weeks gestation, with a birth weight of 3100 g (25th centile) and a birth length of 49 cm (25th centile). Pregnancy and delivery were uneventful. She was the second daughter of healthy, non-consanguineous parents. In the neonatal period hypotonia and feeding problems were present. The anterior fontanel was enlarged and closed at the age of 2 years.
Key points
-
We have cloned the breakpoints of a de novo translocation t(X;9)(p11.23;q34.3) in a mentally retarded female. The translocation disrupts the gene ZNF81 on the X chromosome.
-
ZNF81 is highly homologous to ZNF41, another zinc finger gene associated with non-specific X-linked mental retardation.
-
Mutation analysis in more than 300 families and patients with non-specific X-linked mental retardation revealed a sequence change p.S179N in MRX45, fully segregating with the phenotype.
-
It is likely that ZNF81 represents a novel MRX gene which is causally involved in a minority of patients with non-specific X-linked mental retardation.
Psychomotor development was delayed and she walked at the age of 2 years and 6 months, with hardly any speech development. She had a mild-moderate conductive hearing loss on both sides. At the age of 2 years, an atrial septum defect type II with a pulmonary stenosis was diagnosed without haemodynamic consequence.
At the age of 4 years, she had epileptic convulsions with EEG abnormalities, but thereafter no more convulsions occurred and EEG patterns returned to normal. She had severe behavioural problems involving autistic features, aggressiveness, and auto-mutilation. The menarche started at 10 years of age. At the age of 11 years, she was a non-cooperative, mildly obese girl with apparently normal height and head circumference, brachycephalic skull and several facial dysmorphisms: synophrys, upward slanting of the palpebral fissures, hypertelorism, a broad nasal bridge, protrusion of the tongue, eversion of the lower lip, and small ears.
XLMR families and controls
DNA from approximately 300 families collected by the European XLMR Consortium, including 24 families with linkage to the Xp11.23 region (LOD score >2), were available for mutation screening (www.euromrx.com).
DNA from 241 Caucasian control males and 105 control females was available to verify the incidence of nucleotide changes. In addition, 217 MR males negative for FMR1 trinucleotide expansions were also analysed for mutations in the ZNF81 gene.
Fluorescence in situ hybridisation (FISH)
BACs on chromosome Xp11.2 and 9q34.3 were selected from the UCSC Genome browser (http://genome.ucsc.edu) and obtained from the Max Planck Institute, Berlin. DNA from BACs was labelled with digoxigenin-dUTP by nick translation (Invitrogen) or by random priming with biotine-dNTP by Klenow polymerase (Roche). The hybridisation signals were made visible by antibody-FITC or by avidine-Cy3 labelled antibodies on a Leica DMRA fluorescence microscope.
Southern blotting
DNA samples of cell lines of the patient, one control female, X chromosome hamster somatic hybrid, and hamster, were digested with five different restriction endonucleases (BamHI, BglII, EcoRI, HindIII, SmaI), electrophoresed on (0.6%) agarose gels and transferred in 1.5 M NaCl/0.5 N NaOH to nylon membranes (Genescreen) according to standard protocols.
cDNA fragments or genomic DNA templates were used to generate probes by extension of random hexamer primers with Klenow polymerase in the presence of [32P]dCTP. Probes were hybridised to the Southern blots in 0.125 M PO4, 0.25 M NaCl, 1 mM EDTA, 7% (w/v) SDS, 10% (w/v) PEG 6000 overnight at 65°C.
Blots were washed in 40 mM NaPO4/0.5% SDS at 65°C and exposed to radiographic film.
Vectorette PCR
A vectorette-PCR cloning procedure was used essentially as described by the manufacturer at www.genosys.com. Briefly, BamH1 digested DNA from the patient and a control was ligated to a phosphorylated BamH1-vectorette top strand primer (VTSP-BamHI; 5′-GATCCAAGGAGAGGACGCTGGTCTGTCGAAGGTAAGGAACGAACGAGAGAAGGGAGAG-3′) and a phosphorylated vectorette bottom strand primer (VBSP; 5′-CTCTCCCTTCTCGAATCGTAACCGTTCGTACGAGAATCGCTGTCCCTCTCCTTG-3′) o/n at 16°C. The resulting vectorette libraries were amplified using a universal primer (UVP; 5′-CGAATCGTAACCGTTCGTACGAGAATCGCT-3′) and gene-specific primers (reverse) in exon 6 (5′-CGTGGTGACTATAAGAAGAGTTC-3′ and 5′-GGGAAGGCTTAATTCCAAATTTCC-3′). The latter was used in a semi-nested PCR reaction. Primer positions are indicated in fig 1A. The PCR conditions were as follows: five cycles of 30 s at 94°C, 3 min at 70°C; five cycles of 30 s at 94°C, 3 min at 68°C; 25 cycles of 30 s at 94°C, 1 min at 66°C, 2 min at 70°C. PCR products were purified from agarose gel using a Qiaquick gel extraction kit (Qiagen) and subsequently sequenced on an Applied Biosystems 3730 automated sequencer.
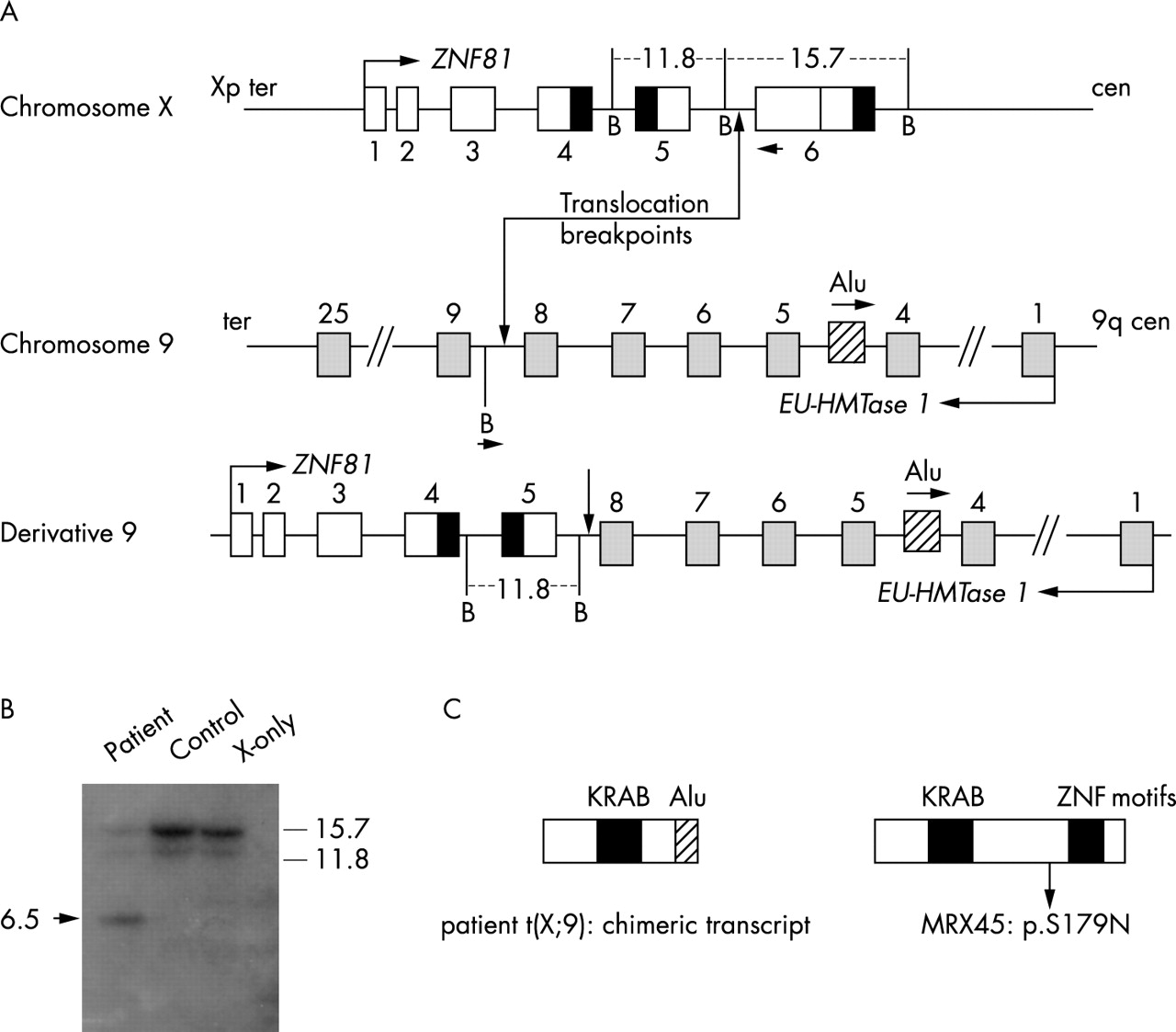
(A) Schematic diagram (not to scale) indicating the exon–intron structures of ZNF81 and Euchromatic histone methyltransferase 1 (Eu-HMTase1) relative to the breakpoint region. Also shown is the der(9) chromosome from which the chimeric ZNF81-Alu transcript is generated. The der(X) chromosome, which was used as a template for the vectorette PCR, is not shown. B, BamHI restriction site. The vectorette PCR was designed with a reverse primer in exon 6 of ZNF81 and a BamHI universal linker primer. The positions of both primers resulting in the vectorette PCR product are indicated with an arrow head. (B) Southern blot of genomic DNA of the patient, control, and X-only somatic cell hybrid digested with BamHI. The probe used corresponds to the cDNA sequence of exon 5–6 of ZNF81. The aberrant fragment specific to patient DNA (arrow head) corresponds to approximately 6.5 kb. (C) Schematic diagram of the chimeric product in the translocation patient and the position of the p.S179N in MRX45. In the chimeric transcript, the 3′ end of exon 5 of ZNF81 is fused to an Alu repeat which is located in intron 4 of Eu-HMTase1 in the reverse orientation. The sequence change in MRX45 is in the 5′ end of exon 6 of ZNF81.
RNA isolation and expression studies
RNA was isolated from patient and control lymphoblastoid cells using an RNeasy kit (Qiagen) according to the instructions of the manufacturer. cDNA was synthesised from 250 ng total mRNA using random hexanucleotide primers and M-MLV reverse transcriptase (RT; Invitrogen). RT-PCR was performed to amplify ZNF81-derived transcripts in cells from the patient and control. For this, specific primers were selected for exons 1, 3, and 6 of ZNF81. A 3′ RACE experiment on the patients RNA was carried out according to the manufacturers protocol (Invitrogen). Primer sequences are available upon request.
Mutation screening in XLMR probands
The entire coding region of the ZNF81 gene was sequenced in 536 MR patients, that is 319 from the European XLMR Consortium and 217 MR males negative for FMR1 mutations, using genomic DNA as a template. Primers were designed to amplify the non-coding exons 1 and 2 and coding exons 3–6 of the gene. Exon 6 was amplified in three overlapping fragments (6a,b,c). Primers and PCR conditions are shown in table 1. The PCR samples were directly sequenced, after purification with a Qiagen PCR purification kit, on an Applied Biosystems 3730 automated sequencer using the same primers as in the PCR.
Primers and PCR conditions for direct sequencing of coding region of ZNF81
RESULTS
Mapping of chromosome Xp11.23 and chromosome 9q34.3 breakpoints using FISH
Successive fluorescent in situ hybridisation with nine overlapping BAC clones showed that RP11-1145B22 gave signals on the derivative chromosome X as well as on the derivative chromosome 9 (fig 2), which indicated that the breakpoint region on the X chromosome was located within this clone. The overlapping neighbouring clones, RP3-393P12 at the distal side and RP11-1154B20 at the proximal side, gave signals on only one of the translocation chromosomes, the derivative-9 and derivative-X, respectively (data not shown). It thus seemed that the X-chromosomal breakpoint is located in a 40 kb fragment that is unique to RP11-1145B22.

Patient metaphase chromosomes were used for fluorescence in situ hybridisation of X-chromosomal breakpoint spanning BAC clone RP11-1145B22 on the normal X, and a split signal was obtained on the der(X) and der(9) as indicated.
In silico analysis
In silico analysis using the UCSC genome browser, http://genome.ucsc.edu/cgi-bin/hgGateway (accessed in March 2004), revealed several spliced ESTs but no known genes, mapping to the breakpoint region. The spliced ESTs did not have a long open reading frame (ORF). However, several of these ESTs showed homology to KRAB-domain coding sequences, which are commonly found in zinc finger proteins. Further BLAST searches with sequences from BAC RP11-1145B22 resulted in the identification of sequences encoding zinc finger motifs that had the same orientation as the KRAB-domain sequences. It appeared that merging of six of the predicted exons of the region had the capacity to form a transcript with an ORF of 1986 bp (661 amino acids). Part of this predicted transcript had been deposited previously in the GenBank database as the zinc finger gene ZNF81 (accession number P51508). The full length transcript has been deposited in the GenBank database with accession number AY487248.
Full-length ZNF81 transcript is absent in the patient cell line
RT-PCR was performed on RNA from cell lines of the patient, a control, and hamster somatic cell hybrids that contained either an active (Xa) or an inactive (Xi) human X chromosome.
Products of the expected sizes were obtained by RT-PCR amplification from exons 1–3 and from exons 1–5 in the control and in the Xa hybrid. No RT-PCR product could be amplified from the Xi cell line, indicating that the ZNF81 gene is subject to X-inactivation. Sequence analysis confirmed the proper identity of the RT-PCR products. Surprisingly, identical products were obtained from the patient’s cell line, suggesting that the 5′ end of the ZNF81 gene is transcribed from the der(9) chromosome. RT-PCR amplification from exons 5–6 was unsuccessful in the patient and in the Xi, whereas a normal product was obtained from the Xa and control cell lines. This result suggested that the translocation breakpoint was located between exons 5 and 6 (fig 3).

RT-PCR for ZNF81 in somatic hybrid cell lines with active X chromosome (Xa) and with inactive X chromosome (Xi) and in patient and control cell lines. Transcripts in the patient were obtained from exons 1–5, but not from 5–6. The (+) and (−) indicate the use of reverse transcriptase.
To elucidate the nature of the der(9)-derived ZNF81 transcript, a 3′ RACE experiment was performed. A single 3′ RACE product was obtained, which appeared to be derived from a chimaeric transcript consisting of the 5′ end of ZNF81 fused to a poly(A)-containing Alu repeat element on chromosome 9. The Alu repeat is fused to the last nucleotide of exon 5 of ZNF81, which indicates that the X-chromosomal breakpoint is likely located in intron 5 of the gene (fig 1C).
Identification of the X chromosome breakpoint by Southern hybridisation
Southern blot analysis was performed to narrow down the position of the X-chromosomal breakpoint. An aberrant restriction fragment of 6.5 kb was observed in the patient BamHI digest with the probe corresponding to the cDNA sequence of exon 5–6 of ZNF81 (fig 1B). The BglII digest of the patient showed a 6.7 kb band of increased intensity as compared to the control, probably caused by the hybridisation of an aberrant restriction fragment of the breakpoint chromosome in combination with a normal restriction fragment of the wildtype allele of the same size.
Another hybridisation with a probe containing part of exon 6 confirmed that the aberrant 6.5 kb band observed in the first hybridisation in the BamHI digest of patient DNA was derived from the normal 15.7 kb band (data not shown). These findings resulted in a reduction of the genomic breakpoint region on the X chromosome to a 816 bp region in intron 5 of ZNF81.
Vectorette PCR
A vectorette PCR was performed in order to determine the exact sequences at the chromosomal breakpoints. To this end, genomic DNA from the patient was digested with BamHI and ligated with a vectorette linker. A PCR reaction was then performed using a universal vectorette primer and a primer specific for exon 6 of the ZNF81 gene. The product obtained by the vectorette PCR revealed that the breakpoint on genomic level was in intron 5 of ZNF81 and in intron 8 of the gene EU-HMTase1 on chromosome 9 (fig 1A). Additional PCR with primers flanking the breakpoints, exactly located the breakpoint to 817 bp from the 5′ end of exon 6 of ZNF81 and 11 bp from the 3′ end of exon 8 of Eu-HMTase1 on chromosome 9. An intronic deletion of 3 bp was present that could not be assigned nearer to either of the two introns.
Sequence analysis
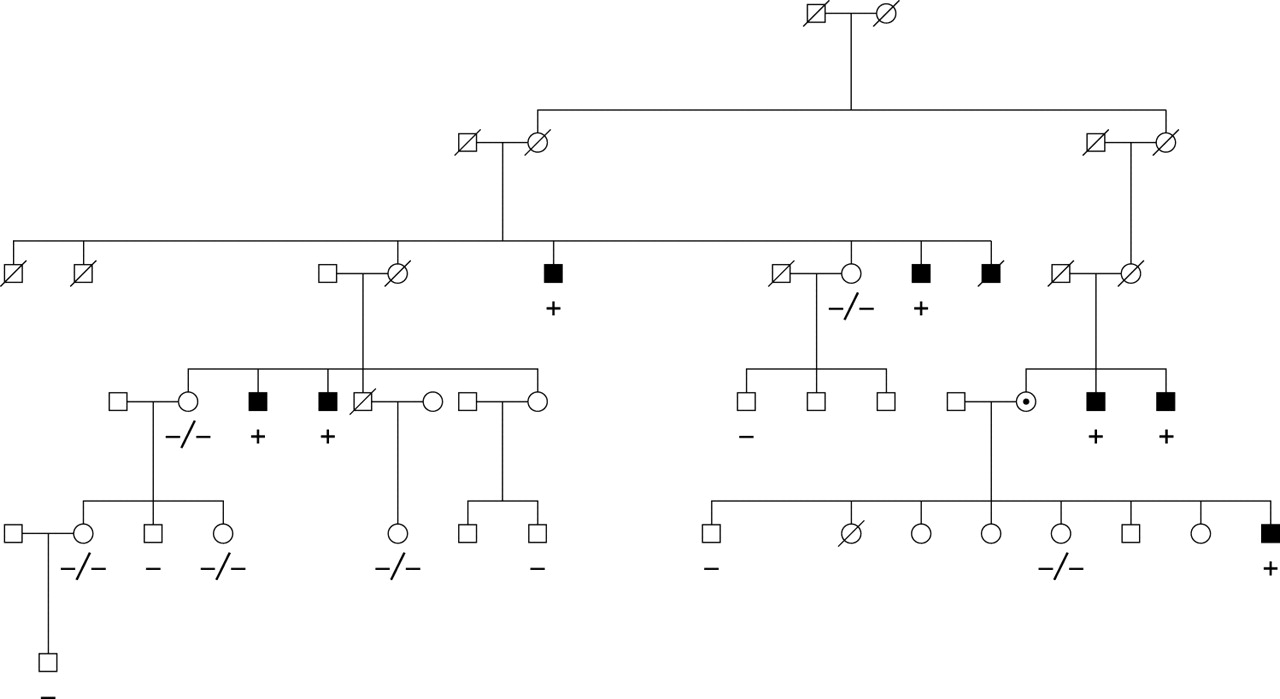
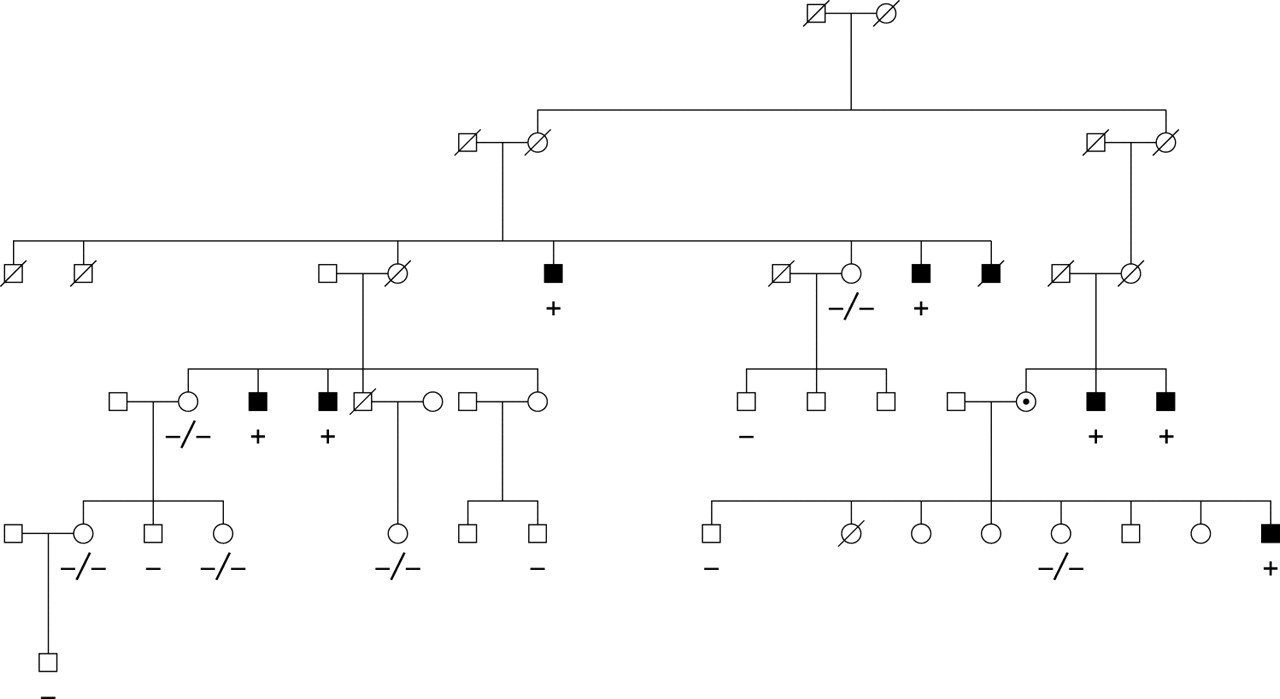
Because Eu-HMTase1 is still expressed in lymphoblasts from the patient, and complete ZNF81 transcripts could not be detected in the cells from the translocation patient, ZNF81 was considered a strong candidate gene for XLMR. Therefore, representative affected males from 24 MRX families mapping to Xp11.2 were screened for mutations in the ZNF81 gene (table 2). In one of these families, family MRX45, we identified a nucleotide change c.536G>A in exon 6, which predicts an amino acid substitution p.S179N. This nucleotide change fully segregated with the phenotype in the family (fig 4) and was not identified in 451 control X chromosomes. One more nucleotide change, c.554C>T (p.S185L), was detected in another linked MRX family, but this change was also found in healthy control males.
Results of mutation screening of ZNF81 in MRX patients
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Segregation of mutation p.S179N in family MRX45.
The mutation analysis was extended to another 285 affected males from unrelated XLMR families and 217 MR males who had been tested negative for an FMR1 mutation. In total one intronic and four exonic sequence alterations were found that were not known as SNPs in genomic databases. However, all of these five changes were also identified in a cohort of 190 control male X chromosomes and 164 female X chromosomes and were therefore considered polymorphisms.
DISCUSSION
We have characterised the chromosomal breakpoints of a mentally retarded female patient with a de novo X;autosome translocation t(X;9)(p11.23;q34.3). The chromosomal breakpoint on the X chromosome of a mentally retarded female patient with a de novo X;autosome translocation t(X;9)(p11.23;q34.3) disrupted the coding region of a Krueppel-type zinc finger gene ZNF81. In the patient only expression of the 5′ end of ZNF81, fused to an Alu element of chromosome 9, was detected. By mutation screening of ZNF81 in families linked to Xp11.2, an sequence change p.S179N was found in MRX45,13 that fully segregated with the phenotype of non-specific MR. Moreover, this mutation was not detected on 451 X chromosomes derived from healthy control individuals. Mutation analysis was extended to 526 MR males, but the five additional sequence changes that were found in this population were all present in healthy controls. The p.S179N though, was only found in MRX45 and not in any of the other 525 MR males, which indicates that the p.S179N alteration is a rare event in the XLMR population. This is not unexpected since in most of the MRX genes mutations have turned out to be very rare.3 This observation suggests that ZNF81 is causative for non-specific X-linked mental retardation, however, the number of controls is not sufficiently large to entire rule out p.S179N being a rare polymorphism.14
Two other mentally retarded female patients with a balanced X;autosome translocation have been reported5,15 where mental retardation appears to be caused by disruption of a zinc finger gene on the X chromosome. In particular, the recent finding by Shoichet et al5 that the highly related ZNF41 is responsible for MRX, strongly supports the notion that ZNF81 is involved in the cause of MRX as well.
As described by Ladomery and Dellaire,16 zinc finger proteins are the most abundant class of proteins in the human proteome. Zinc fingers are ancient motifs capable of mediating interactions with other proteins as well as with DNA and RNA. Almost a third of all Cys2-His2 zinc finger proteins contain a Krueppel-associated box (KRAB) domain, a transcriptional repressor motif that is thought to function through its interaction with the co-repressor KAP-1.17 KAP-1 can interact with the heterochromatin protein HP1 and may mediate transcriptional silencing through the recruitment of histone modifying proteins such as histone methyltransferases and histone deacetylases.18,19 This implies that KRAB/C2-H2 zinc finger proteins are primarily involved in gene regulation. However, it has been shown that some of these proteins play a role in RNA processing as well.20 The multifunctionality of zinc finger proteins is considered to be mediated by alternative splicing and sub-cellular compartmentalisation. At present the functional diversity of the ZNF81 protein is not known. Interestingly, we found that ZNF81 has, at least in lymphoblasts and in fetal brain, one alternative splice product, lacking the non-coding exon 2. We searched for mutations in the 5′ UTR exons 1 and 2, that might have an effect on splicing, but we were not able to identify any such alterations (data not shown).
The mechanism by which the sequence change is responsible for the mental retardation in MRX45 is not clear. The p.S179N is not within a conserved region and it did not have an effect on splicing (data not shown). It is known that the amino acid serine has a potential phosphorylation or glycosylation site. Asparagine does not have such a position, so the p.S179N change might lead to changes in protein folding or structure.
Another interesting point is that four ZNF genes (ZNF41, ZNF21, ZNF157, and ZNF81) are clustered in Xp11.23/p11.3.21,22 It has been postulated that this is the result of duplication events that occurred after evolutionary divergence. Hence, sequence and functional variations in these duplicated proteins may play a role in establishing species-specific attributes.23 It is of note that we were unable to detect any ZNF81-like transcript in mouse brain cDNA using degenerate primers (data not shown). In addition, thorough in silico analysis did not reveal any evidence for a ZNF81 orthologue in mouse, whereas there is one for rat. The same holds true for ZNF41. As it is well known that rats have significantly higher cognitive capacities than mice, it is tempting to speculate that KRAB ZNF proteins may partly account for this. This study and the recent study by Shoichet et al5 have identified ZNF41 and ZNF81 as genes in which mutations are associated with MRX, which would support such role for ZNF genes in cognitive development. The question remains if the disruption of ZNF81 is solely responsible for the phenotype of the proband in this study. Since the breakpoint on chromosome 9q34 interrupts another gene as well, Eu-HMTase1, it is reasonable to speculate that the disruption of this gene may cause some of the dysmorphic features that are associated with the translocation and which are not seen in the patients from the MRX family with non-specific MR. In addition, we cannot formally exclude that disruption of the Eu-HMTase1 gene also contributes to the mental handicap in the translocation patient, which is significantly more severe than the mild MR in the MRX45 family. Further studies on the Eu-HMTase1 gene need to be performed to clarify this.
Acknowledgments
We are grateful to the other members of the XLMR consortium: M Raynaud (Tours), S Lenzner (Berlin), H van Esch (Leuven), and T Bienvenu (Paris).
REFERENCES
Footnotes
-
This work was supported by grants from ZonMw (grant 940-37-031) the Dutch Brain Foundation (grant 10F02(2)43), and by an EU grant (QLG3-CT-2002-01810, Euro-MRX).
-
Conflicts of interest: none declared.