Article Text

Statistics from Altmetric.com
Primary open angle glaucoma (POAG) is the second commonest cause of blind registration in the United Kingdom and affects approximately 70 million people worldwide.1,2 The major risk factors are intraocular pressure, ethnicity, refractive errors, and vascular function. There is also clear evidence from population, case control, and twin studies of a heritable element to POAG.3,4 The genes encoding myocilin and optineurin have been be linked to POAG in large families with autosomal dominant inheritance, and a number of chromosomal loci are being studied for a possible genetic association with glaucoma.5–10 It appears that POAG is a complex trait and multiple genes contribute to the phenotype and increase individuals’ susceptibility to glaucomatous optic neuropathy.
Quite independently, a number of recent studies suggest that apoptosis of the retinal ganglion cells is an important mechanism behind glaucoma.11,12 Apoptosis is a form of genetically controlled, programmed cell death, which is under extensive research, especially in cancer,13–15 neuronal injury,16 and neurodegenerative disorders.17,18 During the primary regulatory steps of apoptosis, a signal of cellular distress activates the tumour suppressor protein p53. This protein acts like a “guardian of the genome”, regulating subsequent apoptotic events through several oncogenes, principally bax and bcl-2.13 Further apoptotic events include changes in mitochondria with cytochrome c release and activation of cystein proteases (caspases), which digest the dying cell from within.19
It is therefore of great interest that one study showed an association between genetic polymorphic variants of the p53 gene and POAG. In a Chinese study, a cytosine (C) residue at codon 72 of the p53 gene was significantly more common in POAG patients than control subjects.20 In another study carried out on an Indian cohort, a second polymorphism (a 16 bp duplication in intron 3 of p53) was also studied, but neither genetic variant was associated with POAG,21 raising questions about their role in POAG. In order to clarify these issues, we studied both polymorphisms in a larger cohort of carefully characterised cases and controls, matched and selected from the same Caucasian population. Rather than focussing on the individual polymorphisms that may not have any direct functional consequences, we studied the p53 haplotype to determine whether natural genetic variants in this gene contribute to the risk of developing POAG.
METHODS
Case and control ascertainment
Having obtained approval from our local research ethics committee, blood samples were analysed from an unrelated Caucasian cohort of 140 POAG patients and 73 healthy individuals, matched for age and gender, from the north east area of England. POAG patients underwent Goldmann tonometry, slit lamp biomicroscopy of the optic discs, Humphrey full threshold 24:2 visual fields, and four mirror gonioscopy. Controls underwent the same examination, omitting gonioscopy and substituting suprathreshold fields for full threshold fields.
Key points
-
Apoptosis of retinal ganglion cells has been implicated in the pathogenesis of primary open angle glaucoma (POAG). Specific genes regulate apoptosis, including p53, which codes for a tumour suppressor protein. Two studies looking at the association of specific polymorphisms in the p53 gene and POAG revealed conflicting results, and the role of p53 in POAG remains uncertain. To address this issue we studied p53 gene haplotypes in a larger group of carefully phenotyped adults with POAG and matched controls from the same Caucasian population.
-
A total of 140 unrelated POAG patients (mean age 73 years, SD 8.01) and 73 unrelated healthy matched controls (mean age 78 years, SD 4.40) were studied. POAG was defined by characteristic cupping of the optic disc, open iridocorneal angle, and typical glaucomatous visual field defects. Secondary causes of glaucoma were excluded, and controls had normal intraocular pressure, visual fields, and optic discs. Patients and controls were genotyped for two p53 polymorphisms (a 16 base pair insertion in intron 3, and a C to G substitution at codon 72 in exon 4 which changes an arginine to a proline residue). Haplotypes were determined using a statistical algorithm.
-
We observed a significant difference in the p53 haplotype distribution between cases and controls (p<0.0001). Subgroup analysis revealed the source of this difference. For individuals with the p53 insertion polymorphism, an arginine at codon 72 was significantly more common in patients than controls (p<0.0001).
-
These findings suggest a potential role for p53 and apoptosis in POAG.
POAG was defined as characteristic cupping of the optic disc, open iridocorneal angle, and typical glaucomatous visual field defects. Patients with secondary types of glaucoma were excluded.
Genomic DNA genotyping
After total genomic DNA extraction, a region of the p53 gene was amplified by polymerase chain reaction (PCR). For each sample, 1 μl of DNA was mixed with 1 U Taq DNA polymerase (Promega), 10× Promega buffer, 2 mmol dNTP, 0.25 μM of each oligonucleotide (primer), and H2O to a total volume of 30 μl. The primers used were: forward: 5′-CCT GAA AAC AAC GTT CTG GTA A -3′; and reverse: 5′-GCA TTG AAG TCT CAT GGA AG -3′. The forward primer was fluorescently labelled at the 5′ end with Dye 3 (Proligo). Cycling conditions were: 94°C for 4 min followed by 35 cycles of 94°C for 1 min, 55°C for 1 min, 72°C for 1 min, and a final incubation of 72°C for 10 min. The full length PCR product was 432 bp in the absence of the 16 bp duplication, and 448 bp in the presence of the 16 bp duplication. A 10 μl sample of each PCR product was digested with 5 U of the restriction enzyme BstUI (New England Biolabs), 2 μl buffer (NEB Buffer 2), and H2O to a total volume of 20 μl per sample for 12 h at 60°C. Restriction fragment length polymorphism analysis was performed with the CEQ 8000 Genetic System Analysis instrument (Beckman Coulter). A 1.5 μl aliquot of the incubated product per sample was added to a mix of 0.5 μl of DNA marker (600 size) and 25 μl of sample loading solution.
DNA fragments with a G at codon 72 (corresponding to an arginine residue) contained the restriction site BstUI, resulting in a fragment of 248 or 232 bp depending on whether the 16 bp duplication was present or absent. DNA fragments with a C at codon 72 (corresponding to a proline residue) lacked the BstUI restriction site, resulting in fragments of 448 or 432 bp depending on the presence or absence of the 16 bp duplication (fig 1). p53 gene haplotypes were determined for the POAG patients and controls using a well-established mathematical algorithm (PHASE version 1.0.1, obtained from the Oxford Mathematical Genetics Group through http://archimedes.well.ox.ac.uk/).22 The frequencies of the four haplotypes were compared using chi-squared analysis. Limited subgroup analyses were performed using Fisher’s exact test.23
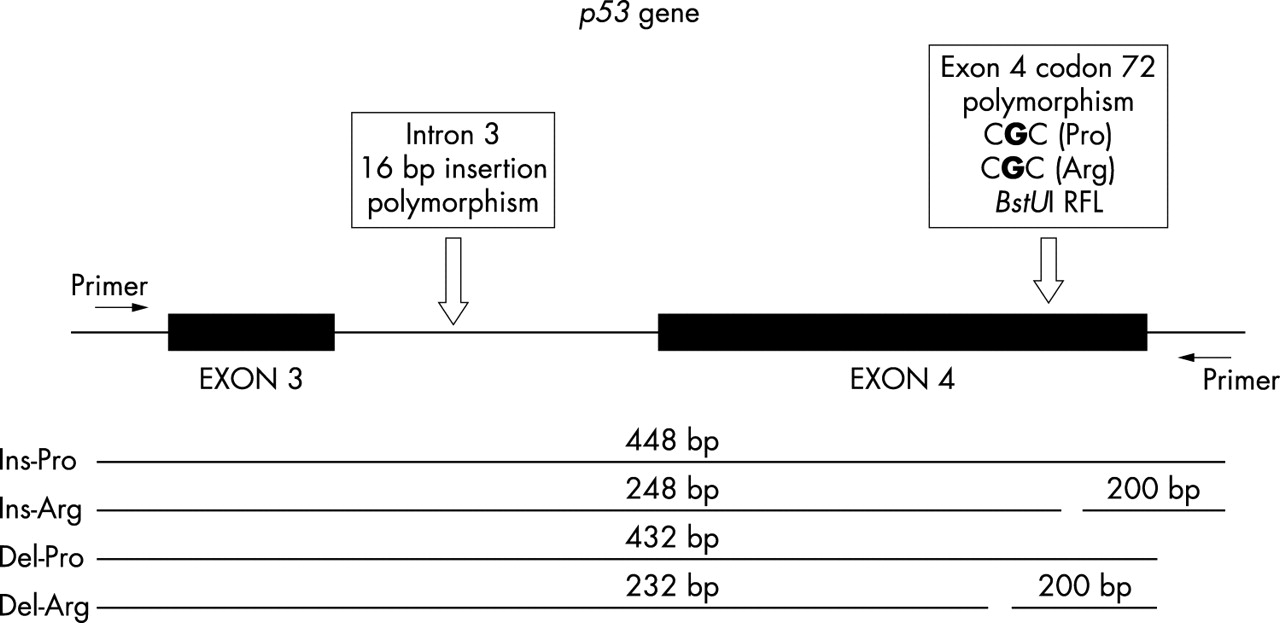
{kind=link}
Diagram showing the amplified area of p53 gene and the studied polymorphic sites (arrows); there is a 16 bp insertion in intron 3 and a base change from C at exon 4 (corresponding to a proline residue) to G (corresponding to an arginine residue, introducing the BstU I restriction site). Schematic representations of the four possible DNA fragments emerging after BstU I digestion are shown. “Ins” and “Del” represent the presence or absence of the 16 bp insertion in intron 3, respectively. “Arg” and “Pro” stand for encoding of arginine and proline from the polymorphic site on exon 4. RFLP, restriction fragment length polymorphism.
RESULTS
Our cohort consisted of 140 POAG patients and 73 controls. The proportion of females in the POAG group was not significantly different from the control group (χ2 = 1.66, p = 0.20). The median age was 73 years for the POAG patients (range 51–87, SD 8.01) and 78 years for the controls (range 68–90, SD 4.4), and the mean age of the controls was significantly greater than that of the POAG patients (Student’s t test = 9.0, p<0.001). Mean intraocular pressure (IOP) was 20.8 mmHg for the patients (SD 2.6) and 16.2 mmHg for the controls (SD 3.4). Median cup:disc ratio was 0.8 and 0.3 for patients and controls, respectively. The frequencies of the four haplotypes are illustrated in table 1.
Frequency distribution of p53 codon 72 polymorphic haplotypes in POAG patients and healthy subjects
Chi-squared analysis revealed a significant difference in the p53 haplotype distribution between cases and controls (χ2 = 37.84, p<0.0001). We then studied the distribution of the haplotypes within the different subgroups. For individuals that did not harbour the p53 duplication polymorphism, the frequency of the G or C residues at codon 72 was not significantly different between cases and controls (Fisher’s exact test, p = 0.15). By contrast, for individuals with the p53 duplication polymorphism, an arginine at codon 72 was significantly more common in patients than controls (Fisher’s exact, p<0.0001). No additional statistical analyses were carried out.
DISCUSSION
POAG is a complex neurodegenerative disease with evidence of a hereditary element. The pathogenetic factors involved in ganglion cell death have not yet been fully explained. Apoptosis has been suggested as a mechanism of glaucomatous neuropathy. The evidence for this hypothesis is based on the presence of characteristic histological and biochemical features in experimental glaucoma and animal studies, including “apoptotic bodies”, DNA fragmentation, and digestion of the dying cells from the surrounding cells, in the absence of inflammatory response.24,25
The p53 gene encodes the tumour suppressor protein p53, which determines crucial events in the apoptotic cascade by regulating other oncogenes. Mutations of the p53 gene, which is located on the short arm of chromosome 17, have been detected in almost 50% of human malignancies.26 The mutant or inactivated p53 protein fails to initiate the apoptotic process and, consequently, genetically damaged cells proliferate in an uncontrolled manner.14 By contrast, in neurodegenerative processes17,18 and toxic neuronal injury,16 the p53 gene is up-regulated in response to cellular stress, thereby promoting cell death through apoptosis. This raises the possibility that functional genetic variants of p53 influence the rate of neuronal cell loss in a number of neurodegenerative disorders, including POAG.
In our study, we have found a highly significant difference in the p53 haplotype distribution between controls and POAG. Subgroup analysis revealed that this was because individuals with the 16 bp insertion in intron 3 were far more likely to have an arginine residue than a proline residue at codon 72 if they had POAG (the insertion-arginine haplotype). These findings add further weight to the growing body of evidence implicating apoptosis in glaucoma, and suggest that genetic variation in the p53 gene is one of the many factors contributing to the aetiology of the disorder.
How can we explain the discrepancy between two previous conflicting reports20,21 and the current study? First, our study was based upon a larger cohort of well-characterised cases and controls. This approach minimises the chance of a false positive result (type I error), to which genetic association studies are particularly prone.27 Second, we cannot exclude the possibility that the different ethnic origins of the study groups are responsible for the disparity in the molecular results. In most cases POAG is thought to arise through a complex interaction of genes with the environment, and different environmental factors, coupled with differences in the genetic background, could alter the relative contribution of p53 polymorphisms in POAG.28 Finally, our analytical approach was fundamentally different to the other two studies. We determined p53 gene haplotypes in cases and controls, and identified a particular p53 haplotype that was strongly associated with POAG. By contrast, the two previous studies independently looked for an association with particular alleles within the p53 gene. This suggests that neither polymorphism is the actual genetic variant that determines the increased risk of POAG, hence the conflicting data in the two previous studies, and that the true functional variant is in linkage disequilibrium with the insertion-arginine haplotype identified in this study. Further analysis of this haplotype will hopefully lead to the identification of the actual DNA sequence that has functional consequences and is responsible for the haplotype association, which may lie in the non-coding regulatory region of the gene.
Acknowledgments
We would like to thank Mr Richard Andrews for his contribution to the above work. PFC is a Wellcome Trust Senior Fellow in Clinical Science.
REFERENCES
Footnotes
-
This study was funded by educational grants from Pharmacia and Special Trustees of the Newcastle upon Tyne Hospitals NHS Trust, UK.
-
Conflict of interest: none declared.
Linked Articles
- Correction