Article Text

Statistics from Altmetric.com
- CFCS, cardio-facio-cutaneous syndrome
- LEOPARD, multiple lentigines, ECG conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness
- ML/LS, multiple lentigines/LEOPARD syndrome
- SSCP, single strand conformation polymorphism
Noonan syndrome (MIM 163950), an autosomal dominant disorder with an estimated prevalence of 1/1000–2500 at birth, is characterised by short stature, facial anomalies, pterygium colli, and congenital heart disease.1,2 Although pulmonary valve stenosis with dysplastic leaflets, hypertrophic cardiomyopathy, and atrial septal defects (ASD) are the most common congenital heart defects in Noonan syndrome,3 a broad spectrum of cardiac phenotypes has been recognised.2–6 About half the affected individuals have PTPN11 gene mutations.7–9 This gene, which maps to chromosome 12q22-qter,10 encodes for the human SH2 domain containing protein tyrosine phosphatase (SHP2).11PTPN11 gene mutations have also been detected in multiple lentigines/LEOPARD syndrome (multiple lentigines, ECG conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness; ML/LS, MIM 151100),12,13 in Noonan-like/multiple giant cell lesion syndrome (MIM 163955),8 in two families presumably affected by cardio-facio-cutaneous syndrome (CFCS, MIM 115150),14 but not in sporadic CFCS.15 The clinical and genetic heterogeneity of these disorders suggests a possible relation between different PTPN11 gene mutations and distinct clinical features. A genotype–phenotype correlation study in Noonan syndrome found an association between pulmonary stenosis and PTPN11 mutations.8 Our aim in this study was to screen a large cohort of patients with Noonan syndrome and ML/LS in order to expand the genotype–phenotype correlation analysis, with particular emphasis on cardiac diseases.
Key points
-
Noonan syndrome and multiple lentigines/LEOPARD syndrome (ML/LS) are associated with a broad spectrum of congenital heart defects in 50–80% of patients. Many patients with Noonan syndrome and most with ML/LS harbour mutations in the PTPN11 gene.
-
The occurrence of PTPN11 gene mutations was investigated in 71 patients with Noonan syndrome and 13 with ML/LS, including 73 with congenital heart defects. A genotype-phenotype correlation analysis was undertaken, with major emphasis on the occurrence of congenital heart defects.
-
Fourteen different PTPN11 mutations were detected in 23 patients with Noonan syndrome and 11 with ML/LS.
-
The distribution of congenital heart defects was markedly different between the two groups. Pulmonary valve stenosis, the most common congenital heart defect in Noonan syndrome, was related to an exon 8 mutation hot spot, while hypertrophic cardiomyopathy, predominant in patients with ML/LS, was associated with mutations in exon 7 and 12. Atrial septal defects were related to exon 3 mutations, while atrioventricular canal defects and mitral valve anomalies were found in association with different exon mutations.
-
Among extracardiac features, only short stature in Noonan syndrome showed a definite relation to PTPN11 gene mutations.
METHODS
Patients
Eighty four white Italian patients were recruited. Informed consent and family history were obtained from all subjects and their parents. Patients were ascertained by clinical geneticists and paediatric cardiologists. Noonan syndrome was diagnosed on the basis of the cardinal features delineated by Allanson1 —in particular short stature, congenital heart defects, broad or webbed neck, chest deformity, developmental delay of variable degree, cryptorchidism, and characteristic facies. For diagnosis of ML/LS, the presence of lentiginosis and café au lait spots was required, in combination with at least three of the cardinal features outlined by Voron et al.16 Clinical assessment included family history, complete physical evaluation of dysmorphisms and malformations, anthropometric measurements, renal ultrasonography, and radiological studies. Standard karyotyping was normal in all patients.
Cardiac evaluation in all patients included preoperative chest x ray, ECG, and cross sectional and colour Doppler echocardiography. Pulmonary stenosis, pulmonary valve leaflet dysplasia, and hypertrophic cardiomyopathy were assessed using classical echocardiographic criteria.2 Cardiac catheterisation with angiocardiography was undertaken in 51 patients as an aid to preoperative assessment, to attempt pulmonary valve dilatation, or to demonstrate left ventricular shape. The cardiac diagnoses were confirmed during cardiac surgery in 32 subjects.
The patients’ parents were examined for major or minor dysmorphisms and cardiac or extracardiac anomalies.
Noonan syndrome was diagnosed in 71 subjects, including one with pigmented villonodular arthrosynovitis, and ML/LS in 13. The study group included 44 females (52%) and 40 males (48%), ranging between 0.1 and 26 years (mean (SD), 8.4 (5.7) years). Seventy four patients were sporadic and 10 familial. One member from each family, presenting with the full blown phenotype, was included in the mutation screening and correlation analysis. Affected and unaffected parents and relatives were screened only after the identification of a PTPN11 gene mutation in the index case.
Mutation screening
PTPN11 gene coding regions and exon-intron boundaries, including about 50 base pairs (bp) upstream and downstream of the exons (GenBank Accession Number: NM_002834), were amplified by polymerase chain reaction (PCR) and analysed by single strand conformation polymorphism (SSCP; Genephor Unit; Amersham Pharmacia Biotech, Uppsala, Sweden). Anomalous fragments were sequenced by the Big Dye Terminator Abi Prism sequencing kit (Applied Biosystems, Foster City, California, USA), and run on an ABI PRISM 3100 genetic analyser automated sequencer (Applied Biosystems). Mutation hot spots were analysed by direct sequencing in patients with Noonan syndrome affected by pulmonary stenosis and in patients with ML/LS not associated with any anomalous mobility shifts on SSCP analysis of exon 8 and exons 7 and 12, respectively, before screening the rest of the gene.
Statistical analysis
All continuous variables were expressed as the mean (SD) for each measurement. Comparisons between patient groups were done using Fisher’s exact test, with a significance threshold of p≤0.05.
RESULTS
PTPN11 mutation analysis
Fourteen different missense mutations, including four novel changes, were detected in 34 patients, including 29 sporadic and five unrelated familial cases (fig 1, table 1). None of these mutations was found in 200 control chromosomes or in unaffected parents (fig 2). In detail, 23 of 71 patients with Noonan syndrome had 12 different PTPN11 mutations, 10 of which were located in the N-SH2 and PTP domains. Codon 308 was a mutation hot spot, while two sporadic mutations were found in exons 1 and 14, at the NH2 and COOH termini, respectively (table 1). Two PTPN11 mutations were found in 11 of 13 ML/LS patients in exon 7 or 12. These mutations were not detected in any of the Noonan syndrome patients.
PTPN11 gene mutations and associated congenital heart defect

The range of PTPN11 gene mutations and associated congenital heart defects in relation to the SHP2 transcript (A) and peptide structure (B). Exon boundaries (exons 1–15) are indicated by white boxes on the bar representing the SHP2 transcript (A). The peptide structure shows SHP2 domains with their amino acid boundaries (B). Noonan syndrome (in bold) and multiple lentigines/LEOPARD syndrome mutations (in italics) are shown, together with the major associated congenital heart defects. Different symbols indicate different types of congenital heart defect. Brackets are used when more than one type of congenital heart defect occurred in the same subject.

{kind=link}
{kind=link}
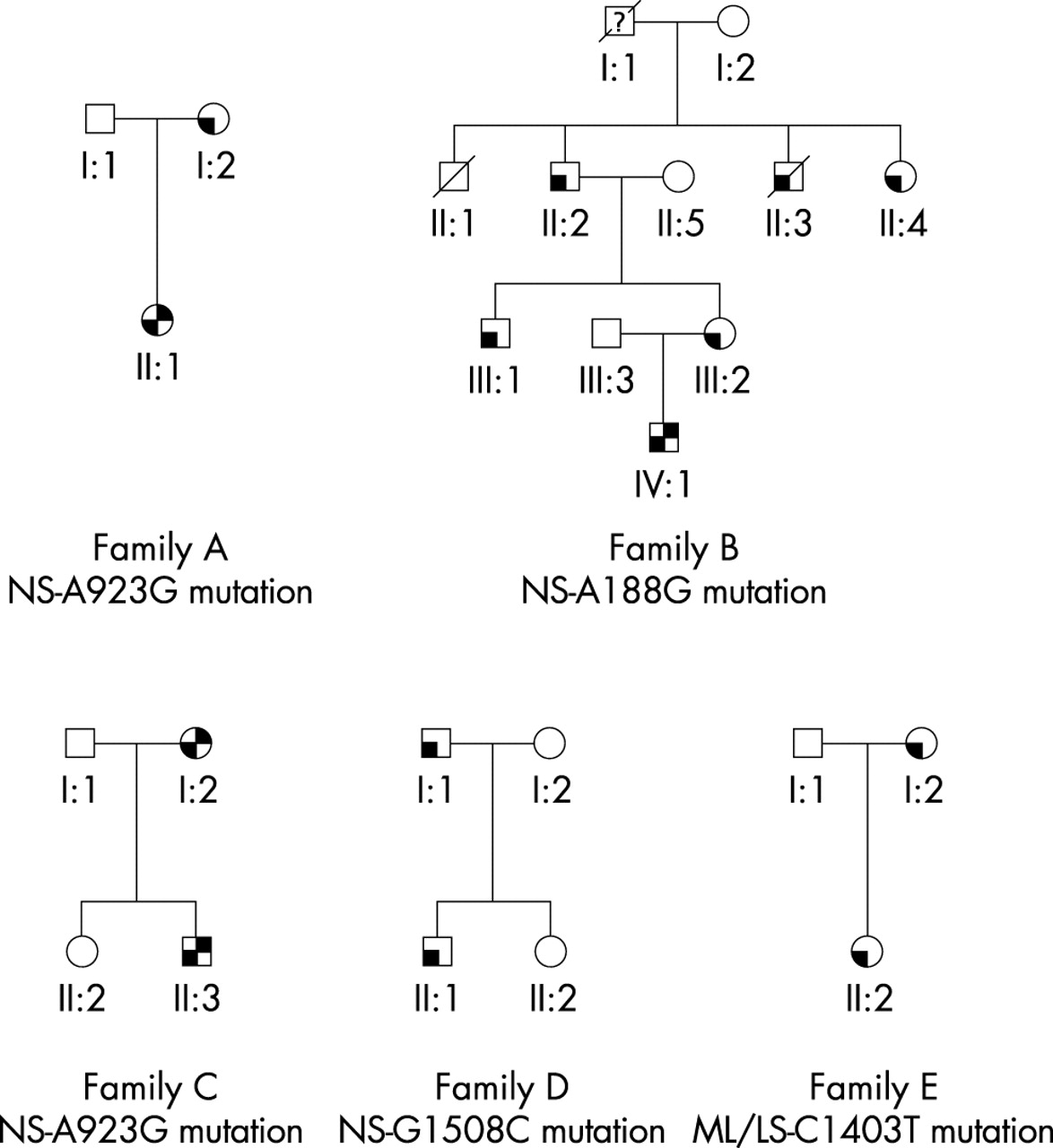
Pedigrees of the families with PTPN11 gene mutations. White symbols represent unaffected family members. Left lower quadrant black symbols represent affected members; left lower quadrant/right upper quadrant black symbols represent affected members with congenital heart disease (pulmonary valve stenosis). ML/LS, multiple lentigines/LEOPARD syndrome family; NS, Noonan syndrome family.
Genotype–phenotype correlation analysis
Twenty seven patients with congenital heart defects (27/73, 37%) had a PTPN11 mutation. The most common congenital heart defects in patients with mutations were pulmonary stenosis (52%) and hypertrophic cardiomyopathy (30%) (table 2). Partial atrioventricular canal defect (AVCD; 11%), ASD (11%), and mitral valve anomalies (MVA; 4%) were also found in association with PTPN11 mutations, while aortic coarctation, tetralogy of Fallot, and ventricular septal defect did not occur in patients with PTPN11 mutations (table 2). Both mutated and non-mutated patients presented a similar spectrum of congenital heart defects. With respect to all 27 patients with congenital heart defects and PTPN11 mutations, pulmonary stenosis showed a significant correlation with mutations in codon 308 (p=0.002), which was a mutation hot spot only in patients with Noonan syndrome. Conversely, hypertrophic cardiomyopathy was more strictly associated with mutations in exons 7 and 12 (p=0.01), a hot spot in ML/LS. The three patients with ASD had mutations in exon 3 (p=0.013) within the N-SH2 domain. Although not statistically significant (p=0.07), a positive trend was noted between mutations in the N-SH2 domain and congenital heart defects. In fact, congenital heart disease occurred in all patients with N-SH2 mutations (11/11), and in only 70% of those with PTP domain mutations (16/23) (table 1). No association was found between AVCD, MVA, and different domain mutations (table 1). Seven of 11 patients without congenital heart defects presented PTPN11 mutations, all within the PTP domain.
Spectrum of congenital heart defect of the 84 patients evaluated in the present study
No significant difference was found in the prevalence of congenital heart defects between Noonan syndrome and ML/LS with PTPN11 mutations (87% (20/23) in Noonan syndrome; 64% (7/11) in ML/LS; p=0.1). Among the Noonan syndrome patients with mutations, pulmonary stenosis was the most common congenital heart defect (65%) and was associated with a codon 308 mutation hot spot (p=0.03), while hypertrophic cardiomyopathy was detected only in the 15% of the individuals with congenital heart defects. Two of three Noonan syndrome patients without congenital heart defects harboured exon 13 mutations. Among the ML/LS patients with mutations, the prevailing congenital heart defect was hypertrophic cardiomyopathy (71%), in association with mutation hot spots in exon 7 and 12, while pulmonary stenosis and AVCD occurred in single patients. Two of four ML/LS patients without structural congenital heart defects had atrioventricular conduction defects (fig 1, table 1).
Among the extracardiac features, only short stature in the Noonan syndrome patients was significantly related to PTPN11 mutations (p=0.04) (table 3). No mutation was found in the only patient with Noonan syndrome who had villonodular arthrosynovitis in the present study.
Extracardiac features of the 84 patients evaluated in the present study
Family analysis
Screening of the PTPN11 gene in 10 familial cases showed mutations co-segregated with the disease in all affected family members in one ML/LS and four Noonan syndrome pedigrees. A concordant congenital heart defect (pulmonary stenosis) was found in a mother and her son with Noonan syndrome carrying the A923G mutation (fig 2, family C). Discordant phenotypes were observed in two Noonan syndrome families (fig 2, families A and B). In family A, the affected mother without congenital heart disease had transmitted the A923G mutation to her affected child with pulmonary stenosis (fig 2, family A). In family B, only one of five affected relatives—all with the A188G mutation—had congenital heart disease (pulmonary stenosis) (fig 2, family B). Two additional families had no congenital heart defects, including a Noonan syndrome father and son pair with the exon 13 G1508C mutation (fig 2, family D), and an ML/LS mother and her daughter pair with the exon 12 C1403T mutation (fig 2, family E).
DISCUSSION
PTPN11 mutations are associated with a broad spectrum of congenital heart defects, both in Noonan syndrome and in ML/LS. This is consistent with animal models, which have suggested a pathogenic role of the SHP2 protein in semilunar valve and myocardial development.17 In the present study we have investigated possible correlations between PTPN11 mutations and clinical features, in particular cardiac defects.
Pulmonary stenosis was the most common congenital heart defect in patients with Noonan syndrome and PTPN11 mutations. However, in contrast to a previous study,8 pulmonary stenosis had a similar frequency in patients with and without mutations. The association between pulmonary stenosis and codon 308 mutations could reflect the presence of a Noonan syndrome mutation hot spot in this position. However, previous genotype–phenotype correlation studies, which recognised the mutation hot spot, did not establish such a correlation. Interestingly, in this cohort of patients, pulmonary stenosis was the only congenital heart defect associated with mutations in codon 308. In patients with PTPN11 mutations, pulmonary stenosis showed a distinct valve leaflet dysplasia (table 2), in agreement with evidence in mice.17 On the other hand, non-syndromic pulmonary stenosis without leaflet dysplasia is not related to PTPN11 mutations,18 suggesting that a different genetic background causes distinct pulmonary stenosis subtypes.19
PTPN11 mutations occurred in eight of 12 patients with hypertrophic cardiomyopathy, without any obvious difference between individuals with and without mutations. The hypertrophic cardiomyopathy rate among ML/LS patients with mutations was higher than previously recognised16 and was clearly related to exon 7 and 12 mutations in the group of patients with mutations as a whole. Interestingly, all ML/LS individuals with hypertrophic cardiomyopathy who have been screened so far harboured a PTPN11 mutation.12,13 Hence, PTPN11 mutations are a major cause of hypertrophic cardiomyopathy in paediatric patients, mainly in the setting of ML/LS. However, the histological features of syndromic and non-syndromic hypertrophic cardiomyopathy are similar,20 and therefore PTPN11 could also be a candidate gene for isolated hypertrophic cardiomyopathy.
ASDs often occur in association with different malformation syndromes.21 Although our cohort of patients with Noonan syndrome and ASD was small, this defect was noticeably correlated with exon 3 mutations.
PTPN11 mutations were associated with partial AVCD in three patients, suggesting that mutations of this gene are responsible of a specific type of syndromic AVCD, similarly to chromosomal aneuploidies (that is, Down’s syndrome and deletion 8p syndrome)22,23 and other gene mutations (EVC gene in Ellis-van Creveld syndrome, DHCR7 in Smith-Lemli-Opitz syndrome, MKS gene in McKusick-Kaufman syndrome, and ZIC3 gene in X linked heterotaxia).24–27 As with ASD, MVA was also associated with an exon 3 mutation. It has been suggested that MVA in Noonan syndrome may be secondary to cardiac jelly and extracellular matrix defects, which are also likely to account for AVCD.3–5,28–30 Hence, myocardial and atrioventricular valve involvement suggests a role for the PTPN11 gene in left ventricular differentiation and mesenchyme development of the atrioventricular cushion.
None of patients harbouring PTPN11 gene mutations had coarctation of the aorta or other common congenital heart defects such as Fallot’s tetralogy or ventricular septal defects. Two ML/LS patients with PTPN11 mutations had conduction defects, which have also been detected in PTPN11 animal models.17 Even if the figure of 87% congenital heart defects in our present series was biased by cardiological recruitment criteria, only 37% of them had PTPN11 mutations. Other gene mutations could account for the heterogeneous cardiac phenotype in Noonan syndrome and ML/LS. Our study has shown that N-SH2 domain mutations are strictly associated with congenital heart defects, although in two Noonan syndrome families not all the affected family members had congenital heart disease. Intrafamilial variability of the cardiac phenotype related to the A188G and A922G mutations points to a reduced penetrance, with a likely regulatory effect of other unknown genes and environmental agents.
The overall PTPN11 mutation detection rate was 40% (39% in sporadic and 50% in familial cases). This figure, which is reduced to 33% if only patients with Noonan syndrome are considered, is slightly lower than previously reported (45%).8 This difference could be explained by assuming that both deletions and missense mutations could have been overlooked by SSCP screening. However, Musante et al have also reported a superimposable rate of mutation in a wide cohort of patients with Noonan syndrome screened by DHPLC analysis.9 On the other hand, 85% of ML/LS patients had PTPN11 gene mutations, with two specific hot spots in the PTP domain. Thus PTPN11 can be considered the major gene of ML/LS.12,13 As previously reported,12 the differential diagnosis between these two syndromes may sometimes be difficult on clinical grounds alone, as specific ML/LS features can appear over the years in subjects diagnosed as having Noonan syndrome. In this respect, it would be of interest to follow up the Noonan patient reported by Tartaglia et al,8 who carried the A836G mutation that was found only in ML/LS individuals in the present and previous cohorts of patients.12,13
In contrast to other reports,8,9 we also found that short stature in Noonan syndrome was significantly associated with PTPN11 mutations, supporting a role of SHP2 in linear growth, as shown by studies in murine models.31
Conclusions
Pulmonary stenosis and hypertrophic cardiomyopathy are the prevailing congenital heart defects in Noonan syndrome and ML/LS associated with PTPN11 mutations. Different heart defects show correlations with the location of mutations within the PTPN11 gene, with statistically significant associations for pulmonary stenosis, hypertrophic cardiomyopathy, and ASD, which appear to cluster to distinct gene domains.
Acknowledgments
This study was supported in part by grants from the Italian Ministry of Health (RC 2002) and from the Italian Ministry of Instruction, University and Research (40% grant to BD). We thank Tania Dottorini and Giorgia Esposito for assistance.
REFERENCES
Footnotes
-
↵* A Sarkozy and E Conti contributed equally to this work.