Article Text

Statistics from Altmetric.com
Neurofibromatosis 2 (NF2), an autosomal dominant disorder that is characterised by tumours of cells of neural crest origin, is caused by inactivating mutations of the NF2 gene on chromosome 22q12.1,2 Bilateral vestibular schwannomas are the most frequent manifestation of the disease, but other central and peripheral nervous system schwannomas, cerebral meningiomas, and ocular abnormalities are also common.3–5 The birth incidence of NF2 is 1 in 33 000 to 1 in 40 000.6 Constitutional NF2 mutations have been found in 30-60% of NF2 patients, and genotype-phenotype correlations have been substantiated.7–12
Half of NF2 patients are new mutations,6 in whom constitutional NF2 mutations can occur either in parental germline cells (prezygotic) or in cells after fertilisation (postzygotic). Postzygotic mutations can result in mosaicism, defined as the presence of a mutation, deletion, or chromosomal abnormality in a group or population of cells.13 Owing to the rarity of NF2, most previous reports of mosaicism in NF2 have been limited in scope,14–16 although a recent study estimated that 25% of NF2 patients with new mutations are mosaic.17 In this study, we describe mutational analysis of 27 mosaic cases of NF2 and the results of genetic testing in their children.
SUBJECTS AND METHODS
NF2 patients were identified through the United Kingdom NF2 registry in the Department of Medical Genetics, St Mary’s Hospital, Manchester. Patients are ascertained by contacting neurosurgeons, otolaryngologists, neurologists, paediatricians, dermatologists, and geneticists throughout the United Kingdom, augmented in the North West Region by the Regional Cancer Registry. There were 297 people from unrelated families who were screened for constitutional NF2 mutations; 256 had bilateral vestibular schwannomas and 41 met other Manchester clinical diagnostic criteria for NF2.3,18 Of these 297 families, 68 families had affected patients in more than one generation, but the founder could be tested in only 27 families.
Genomic DNA was extracted from peripheral blood lymphocytes using standard techniques. DNA was also prepared from fresh or paraffin embedded tumour specimens in 28 NF2 patients, in whom analysis of tumour DNA was necessary to identify the constitutional NF2 mutation. Tumour samples were analysed as previously described.19 DNA samples were amplified for all 17 exons of the NF2 gene using previously described primers.20
Key points
-
Thirty percent of de novo NF2 patients are mosaic.
-
Mosaicism should be suspected in a mildly affected isolated patient with no mutation detected in blood.
-
Risk of transmission to offspring is small in an NF2 patient with a mutation only detectable in tumour.
-
A high risk linkage predictive test in the second generation should be treated with caution.
SSCP/heteroduplex (HA) analysis was carried out on 32 cm long 8% (49:1 acrylamide:bis ratio) native polyacrylamide gels run at 360 V constant voltage for 16 hours at 4°C. SSCP and heteroduplexes were then visualised by silver staining as previously described.21 Samples that gave rise to SSCP or heteroduplex shifts that were present at low levels on silver stained SSCP/HA (minority alleles) were reamplified and loaded onto a second SSCP/HA gel. After electrophoresis, the gel was silver stained as usual, except that the 0.75% Na2CO3 final solution was replaced with 50 mmol/l EDTA. The minority novel bands were then excised from the gel using a fresh sterile scalpel blade. The gel slices were briefly rinsed in 50 μl of TE buffer and the DNA was eluted by crushing and soaking overnight in 50 μl of 1 × TE buffer. The enriched, eluted mutant DNA was then used as the template in a second round PCR, after which the samples were sequenced as described below. PCR products were purified before sequencing using Nucleon QC columns. Sequencing reactions were carried out in both forward and reverse orientations for each sample. Electrophoresis and fluorescent detection of the sequencing reactions were carried out on an Applied Biosystems 377 sequencer with a 48 cm well.
Loss of heterozygosity (LOH) in tumour samples was analysed using three microsatellite markers: D22S275,22 NF2CA3,23 and D22S268.24 NF2CA3 is located within intron 1 of the NF2 gene, and D22S275 and D22S268 are each tightly linked to the NF2 gene (location data base at http://cedar.genetics.soton.ac.uk/public_htlm/).25 LOH was determined using fluorescently labelled PCR product. Lymphocyte-tumour DNA pairs were amplified at the same time and electrophoresed on the same gels. Relative peak heights of lymphocyte and tumour DNA samples were compared using the Genescan version 2.1.1 fragment analysis program (Applied Biosystems). A diminution in relative allele signal strength measured as peak height between lymphocyte and tumour DNA of more than 30% was taken as indicative of LOH.
In addition to SSCP, we used dosage PCR analysis and fluorescence in situ hybridisation (FISH) to detect large deletions because 20-30% of constitutional NF2 mutations are large scale rearrangements.26,27 Dosage analysis was done using a semiquantitative PCR based assay. Four exons from the NF2 gene were amplified in a multiplex with 5′ FAM fluorescently labelled primers (exons 1, 4, 8, and 15) with three control amplicons from the cystic fibrosis gene that were assumed to be present in two copies. Dosage PCR was carried out in 10 μl volumes with 50 ng of genomic DNA. Dosage PCR reactions were carried out for 23 cycles to maintain the amplification reaction within the logarithmic phase. Electrophoresis and quantitative measurement of yield of each product were done using an ABI3I00 Genetic Analyser using Genescan and Genotyper software. Relative peak heights of each test sample were compared to a normalised average of five negative controls using the Dosage Quotient (DQ) method.28 A control deletion positive was included with each run of samples. The acceptable normal range was set as a DQ between 1.15 and 0.85 and the deleted range as a DQ between 0.35 and 0.65.
FISH was carried out using metaphase spreads that were hybridised with three cosmid probes from the NF2 region: 96C10, 10H11, and 121G10. All three probes were derived from the chromosome 22 cosmid library MLL22NC03 (Biomedical Science Division, Lawrence Livermore National Library, Livermore CA). Cosmids 10H11 and 121G10 contain the NF2 gene.2
RESULTS
Mutations were initially identified in 163 (55%) of the 297 families by analysis of lymphocyte DNA. This comprised 58 (85%) of 68 families with affected subjects in more than one generation and in only 104 (45%) of 229 sporadic cases. Twenty further mutations were eventually identified after analysis of tumour DNA from 28/125 of the unfound sporadic cases. One further FISH deletion was identified in lymphocytes from a patient with one of the negative tumours that had been analysed. This gave a final figure of 125 (55%) of 229 families with only one affected subject. It was possible to test the founder subject in 27 of the 68 families with affected subjects in more than one generation; only three (11%) of these founders were mosaics.
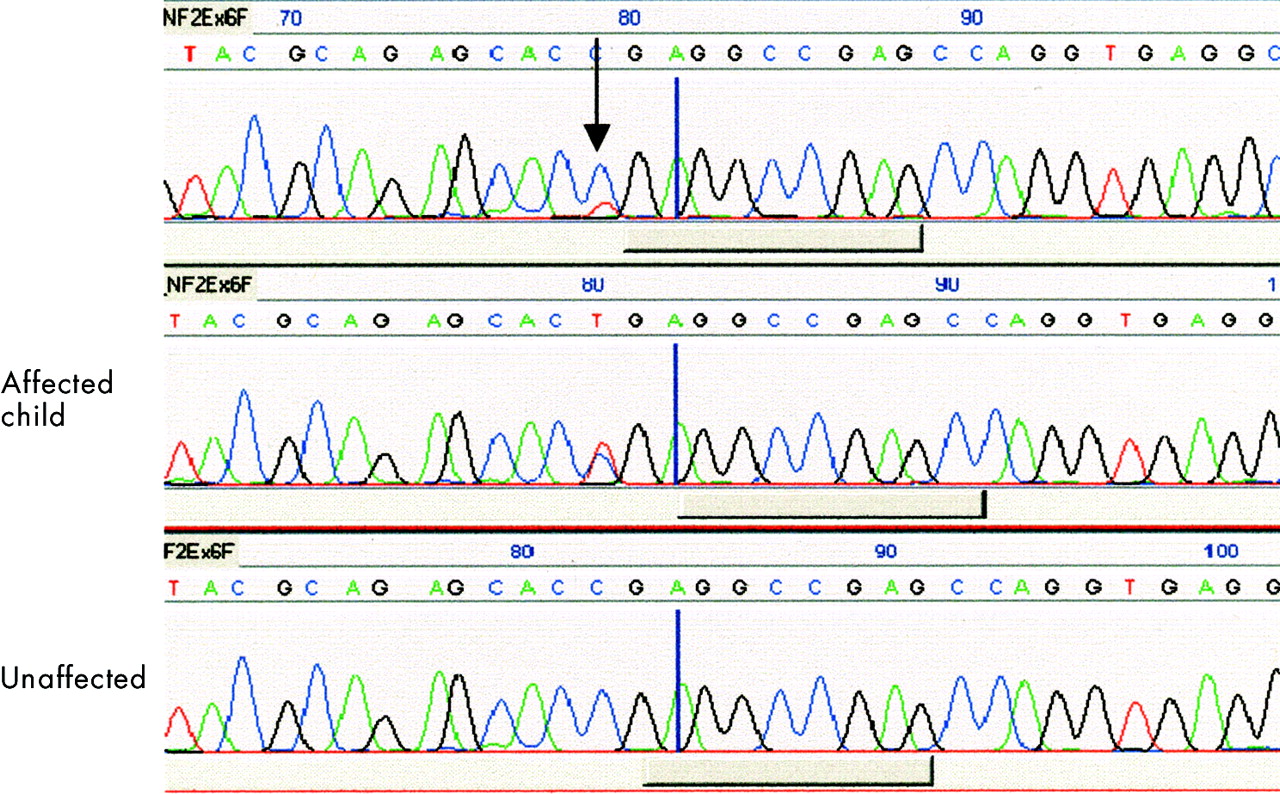
Twenty-seven (9%) of the 297 patients were identified as somatic mosaics by analysis of either lymphocyte DNA or tumour DNA. There were 11 nonsense mutations, 11 frameshift mutations, two splice site mutations, one in frame deletion, one missense mutation, and one large deletion. Ten of the 27 mosaic patients were identified by lymphocyte DNA analysis, as follows. Nine patients had only a weak SSCP/HA shift within their lymphocyte DNA sample, indicating that a minority allele was present. In these patients, conventional DNA sequencing of lymphocyte DNA PCR product often failed to identify a difference from the normal sequence, presumably because the mutant allele was present at too low a level to be detected. However, once the minority bands were excised from SSCP gels, reamplified, and sequenced, a pathological mutation was identified. In two of these nine families, truncating mutations were first identified in blood DNA from a severely affected child, and the mutation was later found in the blood DNA of the parent at lower levels. One family has been reported,16 and another family has a father with a unilateral vestibular schwannoma and two affected children. In both families, a clinically unaffected sib was attributed disease status by linkage analysis, but did not inherit the mutation (fig 1). In a third multi generation family, a deletion was detected on dosage PCR of exon 1 in an affected child of a mildly affected adult male. The deletion could not be detected in the father, but the child was hemizygous for NF2CA3, strongly suggesting that the father was mosaic for the exon 1 deletion. Dosage PCR has not been useful in detecting tumour LOH in our hands, so it is unlikely to detect low level mosaicism (that is, less than 50% of cells carrying the mutation).

Sequence analysis showing a 586C>T nonsense mutation at low level in blood DNA from an affected father, at full level in a severely affected daughter, but absent in a daughter predicted to be affected by linkage analysis.
Seventeen of the 27 mosaic patients were identified through analysis of tumour DNA, as follows. Twenty-eight tumours were analysed from 28 NF2 patients with new mutations in whom a mutation was not found in lymphocyte DNA. Thirteen tumours had both mutational hits identified (LOH as the second hit in 11 tumours), seven had one mutational hit identified, and eight did not have any mutations, deletions, or LOH identified. In 13 patients, mutations were shown to be mosaic by confirming an identical mutation in a second tumour from an anatomically distinct location. In three patients, mutations that had previously been found in tumour analysis were then confirmed at low intensity in blood. In one patient, a multifocal regrowth was found to have the same splice site mutation as the predominant allele with clear loss of the other allele.
One hundred and twenty-five patients with new mutations did not have an identifiable mutation from initial analysis of blood DNA. Tumour analysis was possible in 28 of these people, in whom a causative mutation was identified in 20. Of these 20 people, 17 were mosaic and three had constitutional NF2 mutations at full level in lymphocyte DNA. The remaining 97 people with new mutations did not have tumour analysis.
Mosaicism is more common in NF2 patients with milder disease owing to the smaller proportion of mutation bearing cells.14,15,29 NF2 patients with unfound constitutional mutations tend to have milder disease.4 Therefore, the number of mosaics in the 97 people with new unfound mutations, but who did not have tumour analysis, can be reasonably estimated by extrapolating from the results of tumour analysis, that is, 17 mosaics confirmed at the molecular level (60%) of 28 people with tumour analysis. This proportion, when applied to the 97 people without tumour analysis, yields an estimate of 58 mosaics in this group. Similarly, we found that three (11%) founders in 27 NF2 multi-generation families were mosaic. If this proportion is applied to the 41 multi-generation families in which the founder was unavailable for testing, there would be an estimated four mosaics in this group. The total estimate for the prevalence of somatic mosaicism in the study population of 297 NF2 patients with new mutations is (27 + 58 + 4)/297 = 30%. If the extrapolation to the 41 multi-generation families is excluded, the prevalence of mosaicism is 85/256 = 33%.
We also examined the risk of disease transmission from mosaic parents. Nineteen of the 27 mosaic patients had 45 children; 25 children of 12 parents were tested for the parental mutation (table 1). In 21 of the 25 children who screened negative, the high risk allele could be identified by linkage analysis or from the allele retained in tumour for 14 children. Eight children who screened negative had inherited the high risk allele. Only four of the 25 children (from three affected parents) who were tested had constitutional mutations; three of these children had developed symptoms of NF2 that led to the identification of mosaicism in two of the affected parents.16 The fourth child was diagnosed with bilateral VS on scan; the mildly affected father was inferred to be mosaic for the child’s exon 1 deletion, as described above. The remaining 20 children did not have presymptomatic genetic testing, but none had signs or symptoms of NF2. Four of the 20 children were aged 1-10 years, six were aged 11-20 years, nine were aged 21-30 years, and one was aged 31-40 years. Fourteen of the 20 children had a mosaic parent with a nonsense or frameshift mutation, and seven had a mosaic parent with a splice site mutation.
Description of 27 mosaic NF2 patients
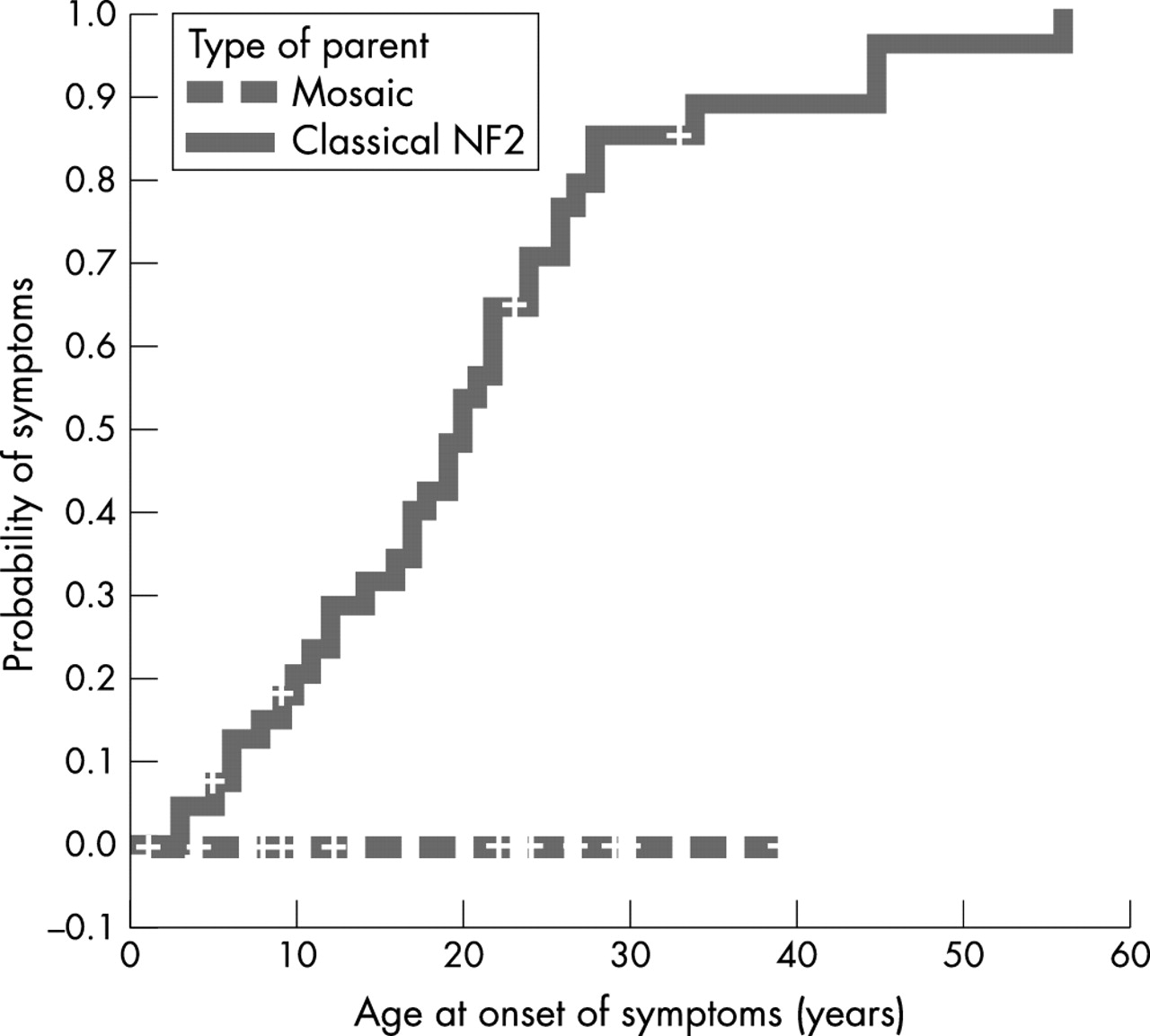
For the presymptomatic children who did not have presymptomatic genetic testing, we used the United Kingdom NF2 registry and Kaplan-Meier analysis to examine the difference in age at onset of symptoms further between these children and children of parents with classical NF2. In the registry, there were 40 inherited cases whose parents had classical NF2 and nonsense or frameshift NF2 mutations, who we compared to the 14 children whose parents were mosaic for nonsense or frameshift NF2 mutations, but who did not have presymptomatic genetic testing. As seen in fig 2, the median age at onset of symptoms in inherited cases whose parents had classical NF2 was 20 years while, as previously noted, none of the 14 children of mosaic parents had developed symptoms (p<0.001, log rank test).
{kind=link}
{kind=link}
Kaplan-Meier analysis comparing the age at onset of symptoms between children of mosaic parents and children of parents with classical NF2 (p<0.001, log rank test). In both groups, the parents have nonsense or frameshift NF2 mutations.
DISCUSSION
In this study, we used a variety of molecular techniques directly to identify 27 somatic mosaics in 297 people with a new NF2 mutation. We estimated that there were 58 mosaics in the 97 patients who had unfound mutations in lymphocyte DNA and in whom tumour analysis was not possible, and that there were four mosaics in the 41 founders of multi-generation families who were unavailable for testing. Taken together with the number of people in whom mosaicism was directly identified, this indicates that 30-33% of NF2 patients with new mutations may be mosaic. This is higher than previous estimates of 15-20%,14,15 and corresponds to the difference in the mutation detection rate between people with new mutations (55%) and inherited cases (85%) in this study. Kluwe et al14 reported direct molecular evidence of mosaicism in six NF2 patients with new mutations by finding mutational hits in blood in four people and in tumour material in two people. The difference in mutation detection rates between people with new mutations and inherited cases was 30%, a difference that they also attributed to mosaicism.
In a recent study that also analysed NF2 mutations in blood and tumours, Kluwe et al17 used a similar approach and estimated that there were 58 mosaics (25%) in 233 NF2 patients with new mutations and bilateral vestibular schwannomas. The results of the present study are consistent with this estimate, and we also assess the risk of disease transmission to offspring. Kluwe et al17 excluded NF2 patients who did not have bilateral vestibular schwannomas, but we have recently shown that patients without bilateral vestibular schwannomas who fulfilled other Manchester clinical diagnostic criteria for NF2 are equally likely to have identified constitutional NF2 mutations.18 In the present study, the prevalence of mosaicism was higher in patients with identified mutations who did not have bilateral vestibular schwannomas, but who fulfilled other Manchester clinical diagnostic criteria for NF2 (nine of 24 patients without affected children, 39%). This may account for the slightly higher estimate of the prevalence of mosaicism in this study than in Kluwe et al.17
The consequences of postzygotic somatic mutations depend on their nature, timing, and location. Loss of function mutations in non-essential or redundant genes probably cause few, if any, consequences in affected cells. As is the case in tumour suppressor genes such as NF2, loss of function mutations that occur in important growth regulating genes and early in development have more severe consequences because cells with a single hit are predisposed to up regulation and tumour growth when a second somatic mutation inactivates the remaining normal copy of the gene. A postzygotic mutation can be passed to the next generation only if it is present in cells that give rise to gametes. Many lethal mutations do not pass to the next generation, as in McCune-Albright syndrome.30 In mice, germline cells separate from somatic cells at a very early stage, suggesting that only very early postzygotic mutations are likely to be represented in somatic cells and the germline.31
A mutation will be passed through the germline but be absent from somatic cells if it occurs in cells that are already committed to gamete formation. Pure germline mosaicism in NF2 has not been reported to date, but suspicion for pure germline mosaicism would be raised if unaffected parents have multiple affected children, since parents with somatic mosaicism are likely to have at least segmental disease, and patients with segmental disease have a reduced likelihood of transmitting disease to the next generation.32 Owing to the rarity of NF2, the probability of unaffected parents having more than one affected child from two independent mutational events is essentially nil, although monozygotic twins with NF2 and unaffected parents have been reported.33 In the present study, no offspring have so far developed NF2 when a parental point mutation was identified in multiple tumours, but not in blood. As such, the risk of disease transmission in this situation is likely to be very small.
In summary, the estimated prevalence of somatic mosaicism in NF2 patients with new mutations is 30-33%, which is higher than previous estimates.14,15,17 The risk of disease transmission from mosaic parents to offspring is very low when the constitutional NF2 mutation cannot be identified in the parent using standard techniques such as SSCP/HA. Somatic mosaicism can cause misleading results in linkage analysis for the second generation,16 and caution should be used in offering presymptomatic testing for at risk subjects in this generation.
Acknowledgments
We thank the many clinicians who sent samples and clinical details of their NF2 patients, in particular Drs Susan Huson, Peter Turnpenny, Jacqui Cook, Eamonn Maher, Ros Ferner, Eric Legius, Andrew Green, and Kathy Tucker. This work was funded in part by the FBT Trust.