Article Text

Statistics from Altmetric.com
Congenital hearing loss has been documented to occur in 1 of 1000 live births, with over half of these cases predicted to be hereditary in nature.1,2 Most hereditary hearing loss is inherited in a recessive manner, accounting for approximately 85% of non-syndromic hearing loss (NSHL). Deafness is an extremely genetically heterogeneous disorder, shown by the fact that 33 loci for recessive NSHL and 39 loci for dominant NSHL have been mapped to date (updated regularly on the Hereditary Hearing Loss Homepage: http://dnalab-www.uia.ac.be/dnalab/hhh/index.html). In populations with increased levels of consanguinity, such as India,3 congenital hearing loss is even higher. According to the 47th Round of the National Sample Survey Organisation (NSSO) taken in 1991 (http://www.healthlibrary.com/), 3 242 000 subjects over the age of 5 have a hearing disability, which is defined as a hearing impairment of 60 decibels and above in the better ear to total loss of hearing in both ears. Prelingual recessively inherited deafness has long been recognised in India. A significant number of the deafness loci have been discovered or found in the Indian population, facilitated by the large extended families and high rates of consanguinity. Seven deafness loci are currently known to be associated with deafness in India. These include DFNB3,4 DFNB5,5 DFNB6,6 DFNB7/B11,7 DFNB15,8 DFNB17,9 and DFNB18.10 In four of these seven cases, the associated gene has been cloned: transmembrane cochlear expressed gene 1 (TMC1) for DFNB7/B11,11 myosin XVA (MYO15A) for DFNB3,4,12 transmembrane inner ear expressed gene (TMIE) for DFNB6,13 and harmonin for DFNB18.14
In many parts of the world, deafness associated with the DFNB1 locus on chromosome 13q11 is the most prevalent. Two genes localised to this chromosomal region have been implicated in deafness, including connexin26 (Cx26, gene symbol GJB2)15 and connexin30 (Cx30, GJB6).16,17 In populations in which the genetic epidemiology of deafness has been evaluated, mutations in GJB2 are the single most frequent cause of inherited deafness. In certain northern European and Mediterranean populations, it accounts for 50% of childhood deafness.18–20 Connexins are gap junction proteins that oligomerise as hexamers to form transmembrane channels called connexons. Connexons from the cell membranes interdigitate to form direct intercellular communications pathways, the gap junction channels. Connexins have a highly conserved form of transmembrane domains separating two extracellular loops from a middle cytoplasmic loop and the N- and C-terminal cytoplasmic ends. In the inner ear, connexin26 is expressed in the supporting cells, stria vascularis, basement membrane, limbus, and spiral prominence of the cochlea.21 The sensory hair cells of cochlea allow potassium ions to pass through during the mechanosensory transduction process of normal hearing. These potassium ions are recycled across the supporting cells and fibrocytes at the base of hair cells through the gap junctions of the stria vascularis and back to the K+ rich endolymph. It is believed that mutations in the GJB2 gene would lead to complete or partial loss of function of the Cx26 protein, interfering with recycling of potassium ions and thus hampering the normal process of hearing. Removal of Cx26 in the epithelial network of the inner ear of mice does lead to deafness in these mice, indicating that Cx26 gap junctions are essential for cochlear function and cell survival.22
Key points
-
Mutations in the connexin26 (GJB2) gene are a major cause of non-syndromic recessive deafness in many parts of the world.
-
To analyse the contribution of connexin26 to non-syndromic hearing loss in India, 215 severe to profound hearing impaired subjects from various parts of the country were analysed. Thirty-eight deaf subjects were found to have biallelic mutations of GJB2. Three different mutations, ivs1(+1)G→A, W24X, and W77X, were found in this study.
-
W24X was found to be the most commonly observed mutation (18.1%). Its carrier frequency was estimated to be 0.024 in a sample of 205 normal hearing subjects analysed. Comparison of the GJB2 flanking markers between W24X homozygotes and a control population suggests the contribution of a founder effect for this mutation.
-
Six gene polymorphisms were observed, of which R127H and V153I were the most common, as well as a novel polymorphism, I111T. This is the first study to report the prevalence of connexin26 and its mutations in the Indian population and has direct implications for genetic counselling in a country with high levels of consanguinity and congenital deafness.
Many studies from various parts of the world have documented the incidence of GJB2 mutations in the deaf population. These include France,19 the United States,23 Israel,24 and, most recently, Lebanon,25 Greece,26 Austria,27 China,28 Brazil,29 and the Iranian30 and Palestinian populations.31 Updates covering all the connexin26 mutations are provided by the Connexin-Deafness Homepage (http://www.crg.es/deafness/). Several of these studies included subjects from the Indian subcontinent.15,19,32–34 However, none of these reports provided a systematic study of the prevalence of connexin26 mutations in the Indian population. In our study, we ascertained 215 profoundly deaf subjects and characterised the GJB2 mutations in this population.
METHODS
Subjects
Independently ascertained probands (n=215) were recruited for the study. Probands with a family history of NSHL had at least two hearing impaired subjects in the family. Sporadic cases of NSHL did not report any known hearing loss in their families and were born to consanguineous marriages. A complete clinical history of each affected subject was collected to ensure that the hearing loss was not a result of infection, acoustic trauma, ototoxic drugs, or premature birth. Ten ml of blood was collected from each subject after written informed consent. This project was approved by the Institutional Bioethics Committee in India. Genomic DNA was extracted using the phenol-chloroform method.35
Audiology
Hearing levels were measured by pure tone audiometry, which included bone conduction. Hearing thresholds were obtained between 250 Hz and 8000 Hz in a soundproof room. All probands exhibited bilateral, severe to profound sensorineural hearing loss.
Connexin26 mutation analysis
The coding exon of GJB2 (exon 2) was amplified with two sets of overlapping primers: 1F (5′-TCT TTT CCA GAG CAA ACC GC-3′) and 1R (5′-GAC ACG AAG ATC AGC TGC AG-3′); 2F (5′-CCA GGC TGC AAG AAC GTG TG-3′) and 2R (5′-GGG CAA TGC GTT AAA CTG GC-3′).24 These two primer sets amplify DNA fragments of 286 bp and 519 bp, respectively. Amplification of the first (non-coding) exon and the flanking donor splicing site was performed by using the Advantage-GC Genomic PCR kit (Clontech) and PCR primers EXON 1A (5′-TCC GTA ACT TTC CCA GTC TCC GAG GGA AGA GG-3′) and EXON 1M (5′-CCC AAG GAC GTG TGT TGG TCC AGC CCC-3′).19 PCR was performed with 150 ng genomic DNA, 25 pmol primers, 10 mmol/l Tris-Cl, 50 mmol/l KCl, 1.5 mmol/l MgCl2, 800 μmol/l dNTPs, and 2.5 U Taq DNA polymerase (Gibco-BRL) in a total reaction volume of 50 μl for 40 cycles (each cycle of 94°C for 30 seconds, 60°C (for 1F-1R primers) and 62°C (2F-2R primers) for 30 seconds and 72°C for 60 seconds). PCR products were purified using Qiaquick columns (Qiagen) and cycle sequencing was performed using 30 ng of the purified PCR product, 3.2 pmol of each primer, and 4 μl of the ABI Prism Bigdye terminator cycle sequencing Ready reaction mix (Perkin Elmer, Applied Biosystems) in a 10 μl final volume for 25 cycles. Following cycle sequencing, the samples were purified with ethanol, precipitated, resuspended in formamide, denatured at 95°C for five minutes and loaded onto an ABI 3100 Genetic Analyzer (Applied Biosystems/Hitachi). The sequence of each amplicon was confirmed by sequencing in both directions. Alignments and analysis were performed using Clustal X version 1.8 (http://www.molbiol.ox.ac.uk/documentation/clustalx/clustalx.html).
Evolutionary analysis
The degree of conservation of the polymorphic residues was analysed using ConSeq (http://conseq.bioinfo.tau.ac.il.), the sequence only variant of Rate4Site, an algorithmic tool for the identification of functional regions in proteins.36 This software examines the closest sequence homologues of a given protein and calculates the evolutionary rate at each amino acid site. Slowly evolving residues are usually important for maintaining the protein’s structure and function. The scores range between 1 and 9, 1 being rapidly evolving (variable) sites and 9 being slowly evolving (evolutionarily conserved) sites.
W24X carrier frequency
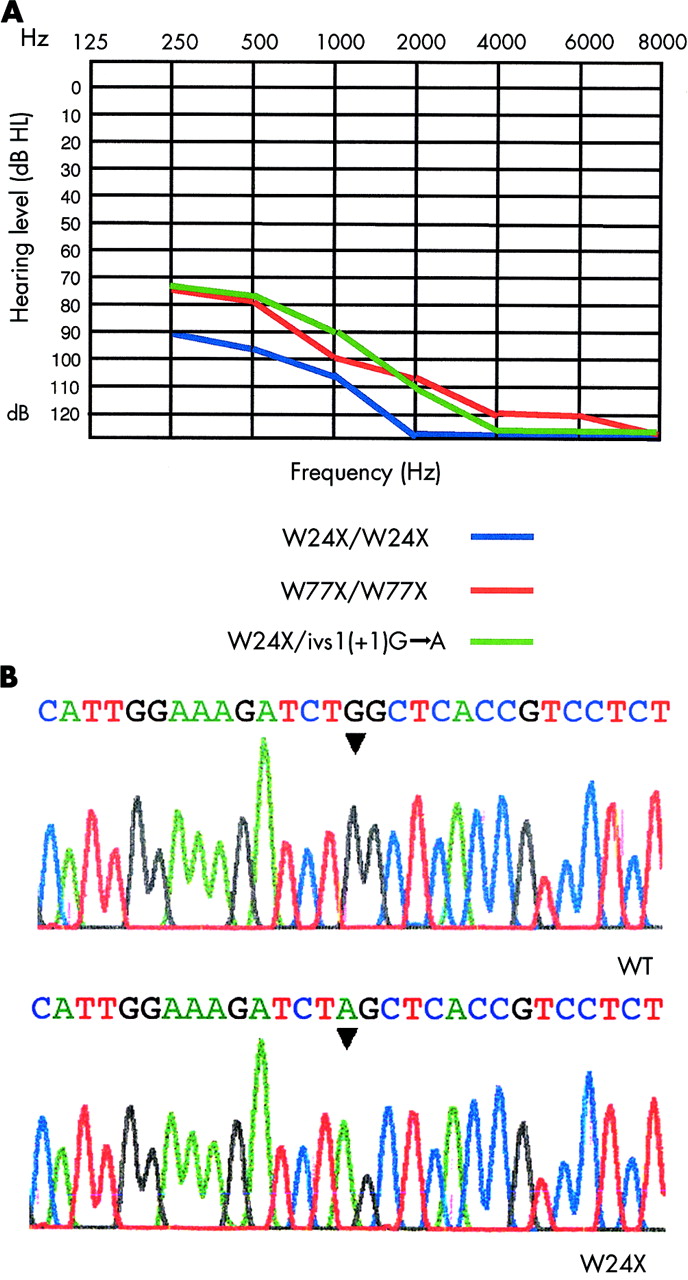
A sample of 205 independently ascertained hearing subjects was used to estimate W24X carrier frequency. To check for presence of the W24X mutation, restriction enzyme digestions were performed on the PCR products obtained using primers 1F and 1R mentioned above. The presence of the W24X mutation introduces an AluI restriction site. AluI digestion of DNA derived from unaffected subjects produced a single fragment of 286 bp, W24X homozygotes produced two fragments of 182 bp and 104 bp, and W24X heterozygotes produced three fragments of 286 bp, 182 bp, and 104 bp.
Mutation age
The age of the W24X mutation was calculated using the equation Pmo−Po= (1−Po)e−ct where Pmo is the frequency of marker allele O on all chromosomes bearing the mutation M, Po is the frequency of marker allele O on all chromosomes in the population, c is the recombination rate per generation, and t is the number of generations.37 Values of the parameters used were: Pmo = 0.780, Po = 0.324, and c = 0.001.
Genotyping
To examine haplotypes associated with the common GJB2 mutations observed, genotyping was performed for the markers D13S141, D13S175, and D13S143 flanking the GJB2 gene. This analysis was performed for the 36 independently ascertained probands who were homozygous for the W24X mutation and in 205 hearing subjects. PCR for the markers was performed using fluorescently labelled reverse primers. Genotyping was done using an ABI 3100 Genetic Analyzer and analysis performed using Genotyper (version 3.7).
RESULTS
Mutation identification
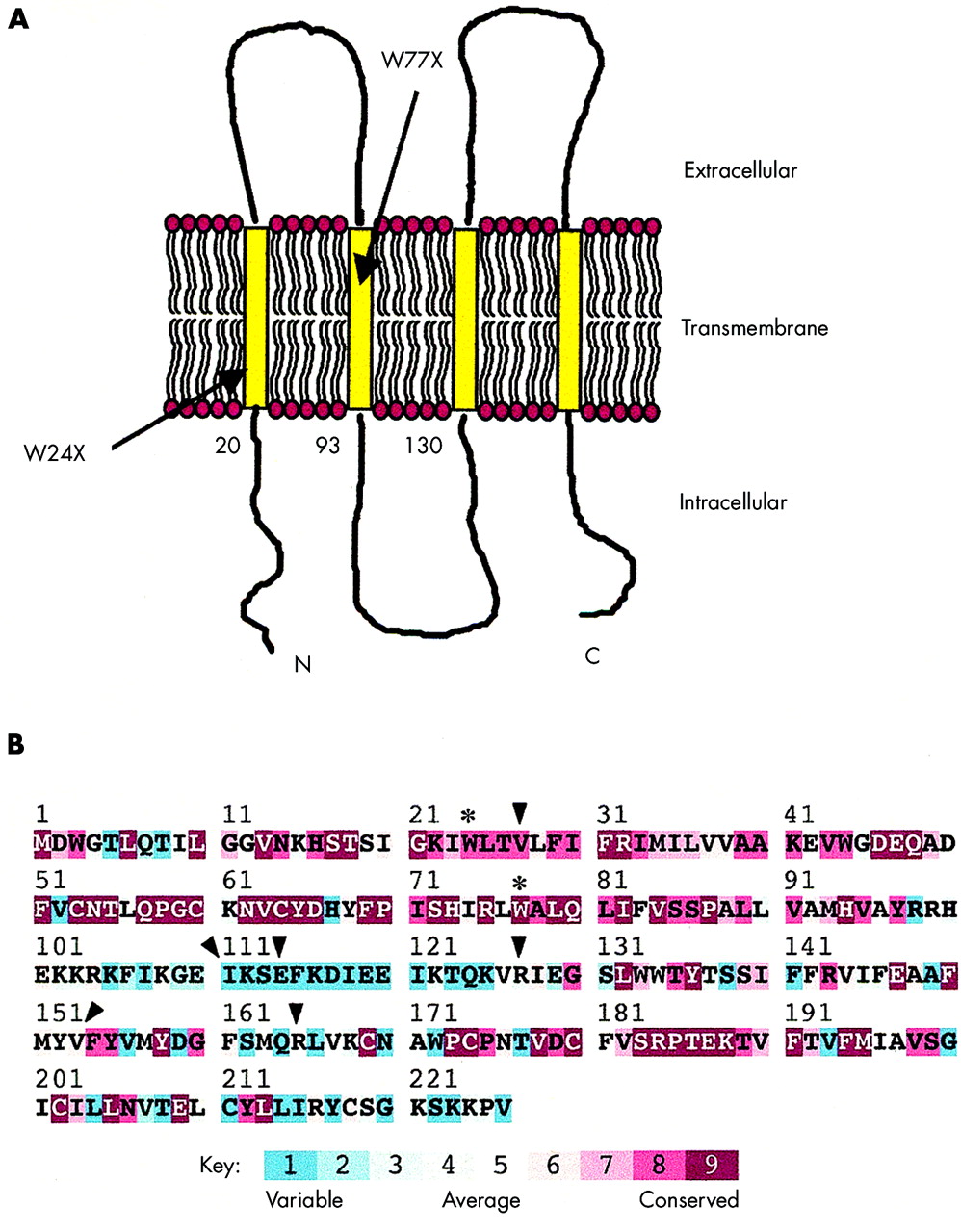
Independently ascertained probands (n=215) from southern and western parts of India were collected from the ages of 1–20 years. Hearing loss was reported to be from birth, without any other accompanying clinical features. The hearing loss was confirmed by audiological testing in a soundproof room. Affected subjects exhibited severe to profound hearing loss in both ears. Fig 1A shows the pure tone hearing thresholds for hearing impaired subjects. The entire coding region of GJB2 was sequenced in the 215 probands. Single affected members were from consanguineous marriages (n=70) and the remainder belonged to families with a history of hearing loss (n=145). A total of three different mutations, ivs1(+1)G→A, W24X, and W77X, were found in this representative Indian population (table 1); ivs1(+1)G→A is a mutation in the splice donor of intron 1, corresponding to −3170 G→A relative to the AUG translation initiating codon. A previous study has indicated that the ivs1(+1)G→A allele is not transcribed or is extremely unstable.31 A G→A transition at nucleotide 71 forms a nonsense mutation at tryptophan 24 (W24X) (fig 1B). W24 is present in the first transmembrane (TM1) domain of Cx26 (fig 2A). Incorporation of a stop codon at this position results in the formation of a protein that is just one-tenth the length of the wild type protein. A G→A transition at nucleotide 231 forms a nonsense mutation at tryptophan 77 (W77X). W77 is present in the second transmembrane (TM2) domain of Cx26 (fig 2A). Incorporation of a stop codon at both these positions is predicted to result in a truncated protein with a complete loss of function.
GJB2 mutations in Indian probands with prelingual, bilateral hearing loss

Connexin26 phenotypes and genotype. (A) Representative hearing thresholds of probands with GJB2 mutations. Only thresholds for the better ear in each case are shown. The horizontal axis shows tone frequency (Hz) and the vertical axis displays hearing level (dB). (B) Sequence analysis of GJB2. The upper panel shows the normal gene sequence and the lower panel shows the sequence of a subject with the W24X/W24X mutation.
{kind=link}
{kind=link}
Connexin26 mutations in India. (A) Schematic representation of the connexin26 protein. The location of the mutations identified are indicated. (B) Amino acid conservation analysis of connexin26. Residue conservation scores, calculated using the ConSeq server from 52 homologous sequences in the Swiss-Prot database,45 are colour coded onto the sequence of connexin26 (see key). W24X and W77X, mutations associated with deafness, are indicated by asterisks; polymorphisms are indicated by arrows.
W24X was the most common mutation seen in the probands; 36 probands were homozygous and six probands were heterozygous for this mutation. One proband was homozygous for the W77X mutation. All the probands who were heterozygous for mutations in GJB2 were typed for mutations in the non-coding exon of GJB2. One of the probands was heterozygous for both W24X and ivs1(+1)G→A. Furthermore, we identified five deaf subjects with a W24X heterozygous mutation; they may be deaf as a result of an unidentified connexin26 mutation or mutations in another gene altogether.
In addition to these deafness causing mutations, six polymorphisms were observed. These were V27I, I111T, E114G, R127H, V153I, and R165W. Their allele frequencies were estimated among the deaf subjects (n=215) and among hearing subjects (n=60) (table 2). R127H was found to be the most common polymorphism among the deaf and hearing subjects (table 2) and was previously identified in one deaf person.38 In this case, an arginine is replaced by histidine in the middle cytoplasmic loop. Another commonly observed polymorphism was V153I, where a valine is replaced by isoleucine in TM3. A novel polymorphism, I111T, was also observed, where an isoleucine is replaced by a threonine. The degree of conservation of the polymorphic residues in Cx26 was analysed using ConSeq, which assigns amino acid conservation grades in the range of 1–9, where 9 is maximal conservation and 1 is minimal conservation, that is, highly variable (fig 2B).
Summary of gene polymorphisms detected and their frequencies
W24X carrier frequency
In order to determine the carrier frequency of the W24X mutation, 205 hearing subjects were screened for this mutation. The presence of the W24X mutation introduces an AluI restriction site. Unaffected subjects, W24X homozygotes, and W24X heterozygotes can be differentiated based on the size of the fragments obtained after digestion with AluI (see Methods). Five out of these 205 subjects were heterozygous for the W24X mutation, implying a carrier frequency of 0.024 (5/205).
W24X founder effect and age of the mutation
To test if the high frequency of the W24X mutant alleles in this sample is a result of a founder effect, we characterised three flanking markers, D13S141, D13S143, and D13S175, in 36 probands homozygous for W24X and analysed the allele frequencies of these markers in 205 controls (table 3). The June 2002 freeze of the UCSC Human Genome Project Working Draft Assembly (http://genome.ucsc.edu/) estimates the distance from D13S141, D13S175, and D13S143 to be 38.5 kb centromeric, 84.7 kb telomeric, and 1.5 Mb telomeric to GJB2, respectively. For the marker D13S141, the 123 allele is the most common in both affected subjects and controls, with no significant difference (χ2=3.49, df 1, p=0.062) in allele frequency (table 3). Similarly, for the marker D13S143, located furthest from GJB2 out of the three, the 126 allele is the most common in both affected subjects and controls, with no significant difference (χ2=0.51, df 1, p=0.475) in allele frequency (table 3). For D13S175, the 100 allele is the most common allele in 36 W24X homozygous probands (allele frequency = 0.78), whereas the frequency of this allele in the control population is lower (allele frequency = 0.324) (table 3). This difference in allele frequency between the affected subjects and controls is statistically significant (χ2=45.62, df 1, p=0.000), suggesting a founder effect for this mutation in the Indian deaf population. The age of the W24X mutation was calculated (see Methods) to be 7880 years (394 generations with each generation of 20 years).
GJB2 flanking markers: allele frequencies among hearing controls and deaf probands with the W24X mutation
DISCUSSION
This study is the first of its kind in India, describing the prevalence of connexin26 mutations in the Indian population. Up to now, studies have included small numbers of subjects from India in the search for causative genes for deafness other than connexin26. The mutations in the coding region (W24X, W77X) described in this report have been found previously in a few subjects from the Indian subcontinent, including India, Pakistan, Bangladesh, and Sri Lanka,15,32,34 whereas ivs1(+1)G→A is reported from India for the first time. We did not find the 35delG or 167delT mutations, which are very common in European and Ashkenazi Jewish populations, with carrier frequencies as high as 2–4%.38–41 Between 35–50% of congenital cases of deafness in southern Europe and the United States have biallelic GJB2 mutations,19,23,38 whereas only 17.7% of congenital deaf probands in India have biallelic GJB2 mutations. Therefore we predict that there will be additional deafness causing genes that are common in India, other than those already found. In a study performed on nuclear, consanguineous families from Tamil Nadu, India, the total number of deafness genes was estimated to be 57.42
We identified several amino acid changes that we defined as polymorphisms since they were present in both the hearing and deaf population. We used Conseq, an algorithmic tool that calculates the evolutionary rate at each amino acid site, to analyse all the polymorphisms. I111T, E114G, R127H, and R165W are predicted to cause no functional changes, since the wild type amino acids at these positions are all variable. Although V27I and V153I are conserved, they too are predicted to be polymorphisms since these are conservative changes. Two recent reports evaluated the function of gap junction channels by transfection of the R127H mutation into HeLa cells. In one case, R127H behaves like wild type connexin26,43 and in the second, while it appears to form gap junctions as observed by immunolocalisation, activity of the gap junction is reduced.44 In the Indian hearing population, R127H was found at a high frequency, strongly suggesting that this is not a causative mutation for deafness.
Among the deaf subjects, five probands were heterozygous for the W24X mutation. The cause of deafness in these subjects could be because of several reasons. The first possibility is that they are deaf as a result of mutations in another gene altogether and they are coincidental carriers. Five of 215 deaf subjects being heterozygous for W24X is not significantly different from the carrier frequency of five out of 205 for the mutation among hearing subjects. The second possibility is the existence of another mutation in the gene’s promoter or unexplored regulatory regions. The third possibility is the implication of another connexin gene (for example, GJB616,17), that is, digenic origin of hearing loss, which could be related to the putative formation of heteromeric connexons or heterotypic channels. The GJB6 deletion was not examined in these cases.
Haplotype analysis of markers flanking the GJB2 gene shows that the most common mutation found in the Indian population, W24X, is most probably a founder effect. While we were unable to make a conclusion regarding the random or founder effect nature of the W24X mutation with the D13S141 and D13S143 flanking markers, results from the D13S175 marker do make this possibility highly likely.
The number of W24X mutations was found to be high, comprising over 95% of the GJB2 mutations found in this sample. The carrier frequency was 0.024, comparable to the carrier frequency of the 35delG and 167delT GJB2 mutations in other populations.38–41 Identifying this mutation in infants would help in the early diagnosis of deafness, so that various intervention strategies could be implemented at an early stage, such as rehabilitation (for example, learning sign language) and fitting of hearing aids, thus avoiding a period of time when the child has little or no auditory stimulation. This mutation can be identified by sequencing and can be easily screened by the restriction enzyme assay we described in a clinical setting. In a country such as India, with high levels of consanguinity and congenital deafness, this assay can be implemented easily and would directly benefit genetic counselling and early rehabilitation in the hearing impaired population.
Acknowledgments
We thank all the subjects who participated in this study. MRS acknowledges a fellowship from the CSIR, New Delhi. This research was supported by the JNCASR, Bangalore and an Indo-Israeli Research Grant from the DBT, New Delhi (AA) and the Israel Ministry of Science, Culture and Sport (KBA). We thank R Narendran, D Gopakumar, Latha Rajendran, Aruna Mahendarkar, Fabian Glaser, and Nir Ben-Tal for help during this work.