Article Text

Statistics from Altmetric.com
The X linked mental retardation group XLMR comprises at least 160 syndromic and non-syndromic disorders of the human X chromosome as summarised by the online catalogue of Mendelian inheritance in man (OMIM; accession date July 2002; http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM). For 113 of these, the gene locus has been mapped, although the gene itself may not have been identified yet.
Börjeson-Forssman-Lehmann syndrome (BFLS)1 belongs to the syndromic forms of XLMR disorders.2 BFLS is best described as a mental deficiency-endocrine disorder. This relatively rare disorder was described for the first time in 1962.1 It is characterised by moderate to severe mental retardation, excessive facial fat, large ears, marked obesity, hypogonadism, and gynaecomastia. A detailed pathoanatomical follow up of the patient from the first publication of the syndrome completed the thorough description of the phenotype.3 Although BFLS is an X chromosomal recessively inherited disorder, it may also be seen in women,2,4 perhaps as a consequence of X inactivation skewing, as has been proven by methylation specific PCR.5 Subjects with BFLS may show intra- and interfamilial phenotypic variability.6 There are other autosomal and X linked disorders with similar symptoms, making the accurate diagnosis of BFLS more difficult. The differential diagnosis of obesity related syndromes includes syndromes like Prader-Willi, Bardet-Biedl, or Wilson-Turner syndromes. However, obesity as a cardinal symptom is not necessarily linked to these loci, as was proven by a linkage analysis of obese sibs.7 Therefore, a thorough analysis of the phenotype is essential. Some features, which are quite frequent in BFLS and allow for the discrimination of this disorder from other mental retardation/obesity related syndromes are: hypogonadism, long, large ears, prominent supraorbital ridge, long philtrum, protruding lips, kyphosis, coarse face, and narrow palpebral fissures, which have been described also as lid ptosis.8
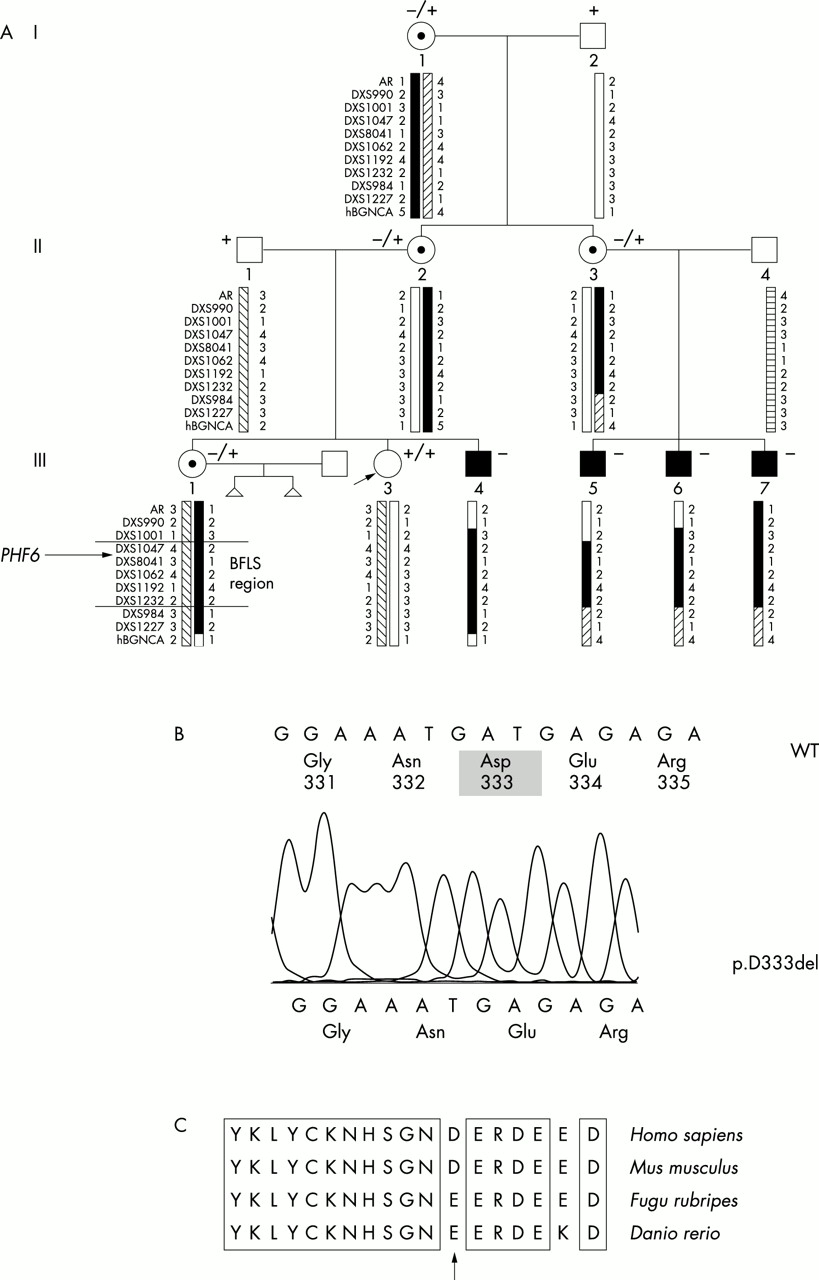
BFLS has been mapped by linkage analysis to the chromosomal region Xq26-q27 with the highest lod score between markers DXS10 and DXS51.9–11 The candidate gene region was refined later by inclusion of additional family members and by an improved genetic map of this region.12 The highest lod score was then calculated for the interval between the markers DXS425 and DXS105. The localisation of the SOX3 gene13 close to the marker DXS51 stimulated the search for mutations in this SRY related HMG box gene. It was found, however, that SOX3 has no mutations in patients with BFLS.12 Meanwhile, a first gene mutated in BFLS has been identified. The zinc finger protein PHF6 was identified by a sequence analysis of ESTs in the BFLS candidate gene region.14 The PHF6 gene (Genbank accession number: GI:27497548) comprises 1095 bp of coding sequence in nine exons. The ORF starts in exon 2 and terminates in exon 10. The mutation analysis of PHF6 in nine families with BFLS showed eight different mutations, two truncation and six missense mutations. Three mutations were detected in exon 2 and one of these, p.C45Y, was detected in two families. In this report we present a new family with BFLS with four affected males and four female carriers. We performed a haplotype analysis in the BFLS interval in order to corroborate the diagnosis of the syndromologist by mapping results. After confirmation of the BFLS locus region, we screened the PHF6 gene for mutations.
Key points
-
Börjeson-Forssman-Lehmann syndrome (BFLS) is an X linked recessive disorder with major symptoms of mental retardation, epileptic seizures, obesity, gynaecomastia, hypogonadism, large, fleshy ears, and prominent supraorbital ridge. The disorder is caused by mutations in the PHF6 gene, which maps to the human Xq26 region. Hitherto, nine mutations have been detected, the majority in exon 2 with one recurrent mutation at p.C45Y.
-
In this report we present a new BFLS family with four affected males and four female carriers. We describe a novel BFLS mutation, c.999_1001delTGA, found in exon 10 of the PHF6 gene. The mutation is a deletion of one triplet in the coding region of the carboxy terminus, which causes deletion of only one amino acid at position 333, p.D333del. It is the only PHF6 deletion mutation identified so far.
-
In order to exclude a frequent polymorphism, 100 control X chromosomes were screened negative for this mutation. Cosegregation of the mutation and the BFLS phenotype was confirmed in the family studied. As a result we conclude that the c.999-1001delTGA mutation is most likely the disease causing mutation in this family.
MATERIALS AND METHODS
Nomenclature
The DNA and protein mutation nomenclature used in this article is in agreement with the recommendations of den Dunnen and Antonarakis.15,16
Patients
All children in our study are from non-consanguineous marriages (fig 1). The index patient III.3 came to the genetic counselling department of Kaunas Medical University. She reported her sister III.1 who had two spontaneous abortions and her affected brother III.4 with mental retardation. In addition, three cousins of hers, all males (III.5-III.7), are also mentally retarded and show similar symptoms. All boys had delayed milestones and none of them had ever attended school. They live today in an institution for severely mentally retarded persons. The characteristic phenotype (obesity, large fleshy ears, gynaecomastia; fig 2) of the brothers and their male cousin prompted our preliminary diagnosis of BFLS. A summary of the symptoms of the affected males of this study is presented in table 1. Informed consent has been obtained from all patients or their representatives in this study.
Clinical signs in the males with BFLS

(A) Haplotypes of the members of the family across the BFLS candidate gene region. Markers outside the region help in pinpointing crossovers. The common affected haplotype found in all affected boys and carrier females (I.1, II.2, II.3, III.1) extends from DXS1047 to DXS1232. These markers define an interval of about 10.5 Mb (BFLS region). The BFLS candidate gene PHF6 maps proximal to marker DXS8041. + and − signs above the symbols for the subjects characterise the sequence variant in exon 10: +, wild type, −, the triplet deletion c.999_1001delTGA. (B) Sequence change caused by the mutation c.999_1001delTGA. (C) Conservation of the amino acid sequence at the p.D333del mutation site. In the pufferfish F rubripes and in the zebrafish D rerio, another acidic amino acid, Glu, is at position 333 (arrow). Sequence identities are boxed.

Subjects III.6 and III.7. The patients have large, fleshy ears. They are obese and show profound gynaecomastia. III.7 has narrow palpebral fissures. Strabismus is obvious in III.7. All other symptoms are summarised in table 1.
Haplotype analysis
DNA was extracted from peripheral blood lymphocytes according to a standard procedure17 and the DNA concentration was adjusted to 100 ng/μl. In order to corroborate our preliminary diagnosis of BFLS and to enable a risk prediction for children of III.3, we performed a haplotype analysis of all family members with markers of the BFLS region. We performed this analysis with 11 highly (heterozygosity ⩾60%, if obtainable) polymorphic microsatellite markers from the BFLS interval (DXS1047, DXS8041, DXS1062, DXS1192, DXS102, DXS1232) and flanking X chromosomal regions in order to detect crossovers. The markers cover the region between proximal Xq and Xq28 in intervals of 600 kb to 21.7 Mb. The markers in the BFLS region map from 125.028 Mb to 135.52 Mb with PHF6 at position 129.461 Mb. Primer sequences were taken from the Genome DataBase (GDB; http://www.gdb.org). Absolute X chromosomal physical map positions (in Mb) of the markers were taken from NCBI’s databases (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Genome). The (CA) dinucleotide repeat sequences were amplified via PCR according to standard PCR protocols (95°C/30 seconds; 52°C-55°C/30 seconds; 72°C/30 seconds; 30–35 cycles; 1.5–2.5 mmol/l MgCl2). The PCR products were separated on denaturing 6% acrylamide gels and alkali blotted onto a nylon membrane. A γ-32P ATP labelled (CA)11 oligonucleotide probe was used as a probe for Southern hybridisation to identify the specific PCR products. The allele sizes of the markers from the pedigree were entered in the Cyrillic Pedigree Analysis software (FamilyGenetix Ltd, Beaconsfield, UK).
Mutation analysis
The coding exons 2–10 of the PHF6 gene were sequenced in genomic DNA of the patients. Exon specific PCR amplification before the sequencing reaction was done according to the previously published protocol.14 The DNA was sequenced by means of a Cy5-AutoRead sequencing kit (Amersham Biosciences) and analysed on an ALFexpress fluorescence sequencing device. In order to check whether the 3 bp deletion in exon 10 which we have detected in this study might be a frequent polymorphism, we screened 100 X chromosomes of unrelated subjects of the central European population with the same ethnic background. For this purpose a PCR with PHF6 exon 10 primers 5′-ATA TAT CAG TGT GTA TTG TAT CC-3′ and 5′-AGG AAT GTT CAT CAG CAG TG-3′ was defined. The resulting PCR fragment measures 244 bp for the wild type sequence and 241 bp for the deletion. The PCR fragments were analysed on an automatic sequencer and the allele sizes were determined with the fragment manager software (Amersham Biosciences).
X inactivation status
X inactivation can be detected by the analysis of the CpG methylation status of the androgen receptor gene AR. The two alleles of this X chromosomal gene in females can be distinguished by size determination of the AR trinucleotide repeat. First, we performed methylation sensitive restriction digests with HpaII from the DNA of the females in order to discriminate for CpG methylation of AR alleles as an indication of X inactivation. Then, two independent PCR reactions with primers flanking the CpG sites and thus also flanking the highly polymorphic trinucleotide repeat of AR were performed on uncleaved DNA and on DNA cut with HpaII. Unmethylated DNA, which is cut by HpaII, will not amplify. Only methylated DNA, which will not be cut by HpaII, will be PCR amplified. The size of the trinucleotide repeat determines the allele size. We measured the peak area of all alleles in the family which allowed us to determine the abundance of active versus inactive alleles as a proof of skewed X inactivation.
RESULTS
We present a three generation family (fig 1) with symptoms of the Börjeson-Forssman-Lehmann syndrome. Both, the phenotype and the X chromosomal mode of inheritance prompted the diagnosis of BFLS. Phenotypic similarities with previously reported cases of BFLS, the comprehensive analysis by a syndromologist, a database search in the Possum database (release 5.2) of the Murdoch Institute (http://www.possum.net.au/), and a database search at OMIM (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM) confirmed the preliminary diagnosis of BFLS and excluded other obesity-mental retardation-hypogonadism syndromes. In order to support this we haplotyped the BFLS region and its flanking regions in all members of the pedigree. We found cosegregation of one haplotype with all affected males and the female carriers (fig 1, 10.5 Mb region from positions 125.046 Mb (DXS1047) to 135.52 Mb (DXS1232)). Recently, the gene implicated in BFLS has been identified to be the PHF6 gene.14PHF6 maps at position 129.461 Mb proximal to the marker DXS8041.
By two point linkage analysis we obtained a maximum lod score of 1.51 at θ=0.00 between the BFLS phenotype and the PHF6 locus DXS8041 at position 129.657 Mb. Formally, this is not a strong support for linkage, but may be a hint to the gene locus.
Since the phenotype and the common haplotype in the candidate gene region both provided evidence for the diagnosis of BFLS, we searched for mutations in the PHF6 gene. Direct DNA sequencing showed a 3 bp deletion c.999_1001delTGA in exon 10 of all BFLS patients. This deletion affects two consecutive codons, but results only in the deletion of one amino acid, the strongly acidic amino acid aspartic acid at position 333, p.D333del. We scanned for this sequence alteration in the entire BFLS family. A 244 bp fragment was obtained in all heterozygous carriers and healthy subjects, whereas the shortened 241 bp was found in all affected subjects and in the heterozygous carriers. As documented in fig 3, it is evident that this deletion was already present in the DNA of somatic cells of the grandmother (I.1). Therefore, females I.1, II.2, II.3, and III.1 are carriers of this deletion. III.1 had two spontaneous abortions and she is a carrier of the mutation. However, it could not be determined whether these fetuses were males and carrying the PHF6 mutation. The index female III.3 does not carry the deletion. To investigate whether this 3 bp deletion represents a frequent polymorphism rather than a mutation, we have screened 100 control X chromosomes. In none of them was a PHF6 deletion identified (data not shown).

{kind=link}
{kind=link}
{kind=link}
Fragment analysis of the PHF6 deletion c.999_1001delTGA in the family with BFLS. The delineation of PCR fragments is the output of the Fragment Manager software. From this analysis it is evident that the mutation was already present in the DNA of the grandmother I.1.
The amino acid sequence flanking the deletion was compared to database entries of other vertebrate sequences in order to check whether the deleted amino acid is conserved in lower species. In mouse the amino acid sequence is identical to the human, whereas the acidic amino acid Asp is replaced by another acidic amino acid Glu in Fugu rubripes and Danio rerio (fig 2C). The flanking amino acids, however, are conserved.
The X inactivation status of all females in our study was examined by a methylation sensitive restriction digest/PCR approach. We found usual ratios of X inactivation in the non-carrier III.3 of 56:44, but also in the carriers of the deletion I.1 with 71del:29, II.2 with 18del:82, II.3 with 39del:61, and III.1 with 68del:32 (where del denotes the allele with the PHF6 triplet deletion). For evaluation of our results, we also included DNA of two females from a recent study14 and measured with our assay inactivation ratios of 98.6:1.4 and 95:5, which confirms skewed X inactivation in these cases.
DISCUSSION
We have studied a three generation family with Börjeson-Forssman-Lehmann syndrome. The family comprised four affected males and four female carriers. The symptoms of the affected boys were somewhat milder than those described by other authors,1,3 making the differential diagnosis more difficult. The gonadotrophins LH and FSH were in the normal range. In addition, hypogonadism in terms of testosterone levels was not markedly pronounced. In comparison to other reports2,8,18 where striking differences in serum testosterone levels in relation to healthy males had been determined, our patients show nearly normal levels, albeit reduced testosterone levels are not a major feature of BFLS. The phenotypic range with overlapping symptoms of other obesity-mental retardation related syndromes may be responsible for the fact that the diagnosis of BFLS is not made more frequently. The phenotype of the subjects from our study (table 1) agrees in most criteria with the major symptoms of BFLS.18 The differential diagnosis includes Prader-Willi syndrome (PWS), Coffin-Lowry syndrome (CLS), and Bardet-Biedl syndrome (BBS1-6). The absence of postaxial polydactyly of both hands and feet, which is a major constituent of Bardet-Biedl syndrome, excludes this disorder in the family of our study. CLS subjects have large, soft hands with tapering fingers, short metacarpals, and drumstick terminal phalanges, but do not show hypogonadism, thus excluding this disorder in our family. We excluded the X chromosomal CLS (and other obesity related syndromes) not by the phenotypic description alone, but also by a haplotype analysis with DNA markers from the BFLS interval and the whole X chromosome (not shown) of affected and unaffected members of the pedigree. Lastly, we also excluded PWS as another obesity mental retardation syndrome based on the large fleshy ears and the prominent supraocular ridge in the affected subjects in our study.
Recently it was described that BFLS is caused by mutations in the zinc finger gene PHF6, making it the first major gene for BFLS.14 We have sequenced the coding region of PHF6 from genomic DNA from our BFLS family and found a 3 bp deletion which leads to the loss of only one amino acid, D at position 333. We have excluded this 3 bp deletion from being a common polymorphism by analysis of 100 X chromosomes of unrelated controls from the same ethnic background. This exclusion is limited to the finding of rather frequent polymorphisms (frequency higher than 1%). The detection of rare polymorphisms (0.1%) with a fairly high power of 95% can only be achieved by studying about 3900 samples.19 There has been only one report on PHF6 mutations in BFLS subjects so far,14 where nine BFLS mutations were reported, four in exon 2, two in exon 7, and one in each of exon 4, exon 8, and exon 10. The majority of the mutations detected were missense mutations with as yet unknown functional consequences. Two nonsense mutations have also been reported leading to PHF6 protein truncation. The c.999_1001delTGA mutation is a novel type of PHF6 mutation. This mutation results in the loss of only one amino acid in a not yet characterised domain of the PHF6 protein.14 The mutation is outside the PHD zinc finger domains and the nuclear localisation sequences (NLS) and lies in a region with unknown biochemical features. The orthologous peptide of other vertebrates is highly conserved in this interval, indicating an indispensable function. Its participation in transcriptional regulation is unclear. In vitro assays with engineered deletions of this part or gene targeting in mice might elucidate the functional consequences of mutations in this part of the gene.
Interestingly, none of the carrier females within this family was affected. This is unusual in view of the recent finding14 where 19 out of 20 unaffected BFLS carrier females tested showed highly skewed X inactivation patterns. When the X inactivation was not highly skewed, mild BFLS features were observed in one carrier female.14 This implies that the c.999_1001delTGA deletion mutation identified in this study may not confer such a selective disadvantage as the other detected PHF6 mutations do, at least based on the blood white cell DNA analysis. In addition, we do not know anything about disadvantages of the p.D333del mutant cell populations during early embryogenesis, where the choice for X inactivation is performed. Hence, a mutation in the functional domains of the protein might be detrimental to the cell if not silenced by X inactivation, whereas the p.D333del mutation might not confer any selective disadvantage. In any instance, this observation may warrant caution when counselling female BFLS carriers.