Article Text

Statistics from Altmetric.com
The Holt-Oram syndrome (OMIM 142900) is an autosomal dominant disorder with clinical features characterised by a variety of skeletal malformations and congenital heart defects.1–5 The gene for Holt-Oram syndrome has been identified as TBX5 on chromosome 12q24.6–11TBX5 encodes a protein of 518 amino acids that belongs to the family of the T box transcriptional factors,10,11 and is expressed in embryonic heart and limb tissues, consistent with its involvement in development of the heart and skeletal structures.12–17 TBX5 contains a highly conserved DNA binding domain, the T box domain. Recently, three groups have provided direct evidence that TBX5 can bind to DNA and activate transcription of its target genes including the gene for atrial natriuretic factor (ANF).17–19 Furthermore, TBX5 can interact directly with the cardiac homeobox protein NKX2.5 synergistically to activate transcription of ANF.17,18
To date, 22 different non-translocation TBX5 mutations have been reported. These include five truncations (nonsense mutations), three splicing changes, six frameshift mutations, seven missense mutations, and one large deletion involving exons 3-9.20–22 Frameshift, splicing, and nonsense mutations are expected to produce truncated TBX5 or no TBX5 at all (for example, via nonsense mediated mRNA decay), which causes Holt-Oram syndrome by haploinsufficiency. However, the mechanisms by which the missense mutations cause Holt-Oram syndrome are not well understood. Ghosh et al19 found that missense mutations G80R and R237Q eliminated DNA binding, whereas mutations G169R and S252I did not affect DNA binding. Hiroi et al18 analysed the effect of G80R and R237Q on transcription activity of the ANF promoter. Mutation G80R caused significant reduction of transcriptional activation activity of TBX5 and loss of synergistic activation with NKX2.5. Surprisingly, mutation R237Q, which eliminated DNA binding, had minor effects on transcriptional activation.18 Continued identification and functional analysis of TBX5 mutations will be invaluable in identifying new mechanisms underlying the pathogenesis of Holt-Oram syndrome.
In this study, we have identified two novel mutations in TBX5, one microdeletion (381-408del27bp) and one nonsense mutation 192G>A (W64X), and report the detailed molecular mechanisms by which the 381-408del27bp deletion causes Holt-Oram syndrome.
MATERIALS AND METHODS
Genotyping, linkage, and mutation analysis
Informed consent was obtained from the participants in accordance with guidelines established by local institutional review boards. The participants involved were evaluated by interviews, physical examinations, x ray, electrocardiography, and echocardiography.
Human genomic DNA was isolated using the DNA Isolation Kits for Mammalian Blood according to the manufacturer’s instructions (Roche Biochemical Inc). Genotyping and linkage analysis were carried out as previously described.23,24
Key points
-
Holt-Oram syndrome is an autosomal dominant disorder characterised by congenital heart defects and upper limb abnormalities.
-
We define here mutations in TBX5, a gene encoding a member of the T box family of transcriptional factors, in affected members of two Holt-Oram syndrome families. A novel nonsense mutation, 192G>A (W64X) in TBX5, was identified in a single patient, and a novel 27 bp in frame deletion (381-408del27bp) cosegregated with 25 patients in a large family.
-
To investigate the mechanism by which the 381-408del27bp deletion causes Holt-Oram syndrome, a TBX5 construct representing the mutation was generated and tested for its abilities to bind DNA and to activate the transcription of a target gene (ANF, atrial natriuretic factor gene). In vitro protein-DNA binding assays indicated that the 381-408del27bp deletion greatly reduced DNA binding ability of TBX5. A transient cell transfection assay showed significantly reduced transcription activation by TBX5 with the 381-408del27bp deletion. Furthermore, the mutant TBX5 did not interfere with the function of wild type TBX5. Immunocytochemistry showed that wild type TBX5 was localised in the nucleus, whereas the 381-408del27bp mutant protein was localised to both nuclei and cytoplasm, with predominant immunostaining in the cytoplasm.
-
These results suggest that impaired nuclear localisation and dysfunction in DNA binding and transcriptional activation are the mechanisms that can account for the loss of functional TBX5 in Holt-Oram syndrome patients with the 381-408del27bp deletion. This study also identified the disruption of TBX5 nuclear localisation as one novel mechanism for Holt-Oram syndrome.
Mutation analysis was performed by direct DNA sequence analysis with an ABI3100 Sequencer. PCR amplification of exons and exon-intron boundaries of TBX5 was carried out as previously described.10,20,25
Plasmid constructs
The upstream region from –270 to +1 bp from the transcription start site of the ANF promoter was PCR amplified and cloned into the pGL3 basic vector (Promega), resulting in the ANF-luc reporter gene.18 The wild type TBX5 expression construct encoding FLAG-tagged TBX5 has been described previously.18 The full length TBX5 cDNA was cloned into the pcDNA3.1 vector (CloneTech).18 The 381-408del27bp deletion was introduced into the WT construct by PCR based, site directed mutagenesis26 and verified by DNA sequencing.
Cell culture, transfection, and reporter assays
Cells were grown in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% fetal bovine serum (Gibco-BRL) to 80-90% confluence, and transfected with Lipofectamine 2000 (Gibco-BRL) and 0.5 μg of DNA for the reporter gene and the expression constructs. The ANF-luciferase reporter gene was cotransfected with the wild type TBX5 expression construct or a mutant construct and 50 ng of internal control plasmid pSV-β-galactosidase (Promega). The total amount of DNA was kept constant at 1.5 μg in each well by adding an appropriate amount of pcDNA3.1 where necessary. Cells were harvested and lysed 24 hours after transfection. Luciferase assays were performed using a Dual-Luciferase assay kit (Promega) according to the manufacture’s instructions. β-galactosidase activity expressed from pSV-β-galactosidase was used to normalise transfection efficiency among wells. The results shown are the mean (SD) from at least three independent experiments and each experiment was performed in triplicate.
In vitro translation of TBX5 proteins and gel retardation experiments
The wild type and mutant TBX5 proteins were prepared using the TNT T7 Quick Coupled Transcription/Translation System according to the manufacture’s instructions (Promega). After the reaction, a mini-protease inhibitor cocktail (Roche Biochemical Inc) was added to the reaction mixture.
For gel shift experiments, the TBX5 consensus core binding site was generated by annealing the oligonucleotide 5′-aataTCACACCTgtac-3′ to the complementary oligonucleotide 5′-gtacAGGTGTGAtatt-3′. The TBX5 binding site was end labelled using T4 polynucleotide kinase (Pharmacia) and γ-32P-ATP, and purified using Sephadex G-30 spin columns (Pharmacia). The 32P labelled TBX5 binding site was incubated with 4 μl of in vitro synthesised TBX5 protein in binding buffer (25 mmol/l Tris-HCl, 20% glycerol, 50 mmol/l NaCl, 5 mmol/l MgCl2, 0.5 mmol/l EDTA, 1 mmol/l DTT, and 1 mg/ml BSA) containing 0.125 μg of poly(dI-dC) in a 20 μl volume at room temperature for 30 minutes. The binding mixture was then fractionated through 6% non-denatured polyacrylamide in 0.5 × TBE buffer at 15 V/cm for 45-60 minutes. After electrophoresis, the gel was dried at 80°C for 40 minutes and exposed to x ray films. For competition experiments, 100-fold excess of cold double stranded competitor oligonucleotides were added to the reaction mixture.
Western blot analysis and immunocytochemistry
Western blot analysis and immunocytochemistry were performed using cells transfected with FLAG tagged wild type or mutant TBX5 constructs as described previously.27
RESULTS
Novel deletion 381-408del27bp of TBX5 in a large family with Holt-Oram syndrome
We studied one large three generation Holt-Oram syndrome family with 48 living family members and 25 affected subjects (kindred HOS001) (fig 1A). Linkage analysis confirmed that kindred HOS001 was linked to TBX5. Two point linkage analysis yielded a lod score of 8.22 for marker D12S1646 and 7.02 at D12S354 at a recombination fraction of zero (TBX5 is located between D12S1646 and D12S354).

Identification of a novel 27 bp TBX5 deletion (381-408del27bp) associated with Holt-Oram syndrome. (A) Cosegregation of deletion 381-408del27bp with the disease in kindred HOS001. Normal subjects are shown as clear circles (females) or squares (males). The symbol with the question mark indicates uncertain phenotype. Symbols with “+”, skeletal and limb abnormalities; symbols with solid left half, ASDs and/or VSDs; symbols with solid right half, bradycardia and other cardiac defects (MVP). The 381-408del27bp allele (Δ27bp) can be easily distinguished from the wild type allele (WT) with a 6% denaturing polyacrylamide gel. (B) Sequence analysis of the WT allele (top) and the 381-408del27bp allele (Δ27bp, bottom). (C) Schematic representation of the TBX5 gene. The TBX5 gene has nine exons (boxes) with exons 3-7 coding for the T box DNA binding domain. The 381-408del27bp deletion is located in exon 5 of TBX5.
To identify the TBX5 mutation in kindred HOS001, we PCR amplified the TBX5 exons from the proband (the second subject in the third generation, fig 1A) and sequenced them. This led to the identification of a novel 27 bp deletion in exon 5 of TBX5 (381-408del27bp) (fig 1B). To confirm the existence of the 381-408del27bp deletion, the PCR product of exon 5 of TBX5 from the HOS proband was separated through agarose gel electrophoresis. Two DNA bands were identified on agarose gels (data not shown), purified, and sequenced directly. This confirmed that the faster moving DNA band contained the 27 bp deletion. This deletion results in a deletion of nine amino acids (K126A127E128P129A130M131P132G133R134) in the T domain of TBX5 (fig 1C).
We then used a denatured polyacrylamide sequencing gel to determine whether all HOS patients in kindred HOS001 carry the 381-408del27bp deletion. As shown in fig 1A, the 27 bp deletion cosegregates with all affected members in kindred HOS001, but not with all normal members. The 381-408del27bp deletion was not observed in DNA samples from more than 200 control subjects (data not shown). These data suggest that the 381-408del27bp deletion in exon 5 of TBX5 causes Holt-Oram syndrome in kindred HOS001.
Novel nonsense mutation 192G>A (W64X) of TBX5 in a patient with Holt-Oram syndrome
We studied a single patient (HOS002) with Holt-Oram syndrome. PCR amplification of TBX5 exons and direct DNA sequence analysis identified a single heterozygous base substitution (C to T or G to A on the complementary strand) in exon 3 of TBX5 (fig 2A, B). This substitution occurs at the second position of codon 64, and changes codon 64 coding for a tryptophan residue into a stop codon (192G>A(W64X)). The existence and nature of the 192G>A(W64X) mutation was confirmed using single strand conformation polymorphism and restriction fragment length polymorphism analyses (data not shown). Mutation 192G>A(W64X) is located in the T domain of TBX5 and truncates the TBX5 protein by 88% (fig 2B).
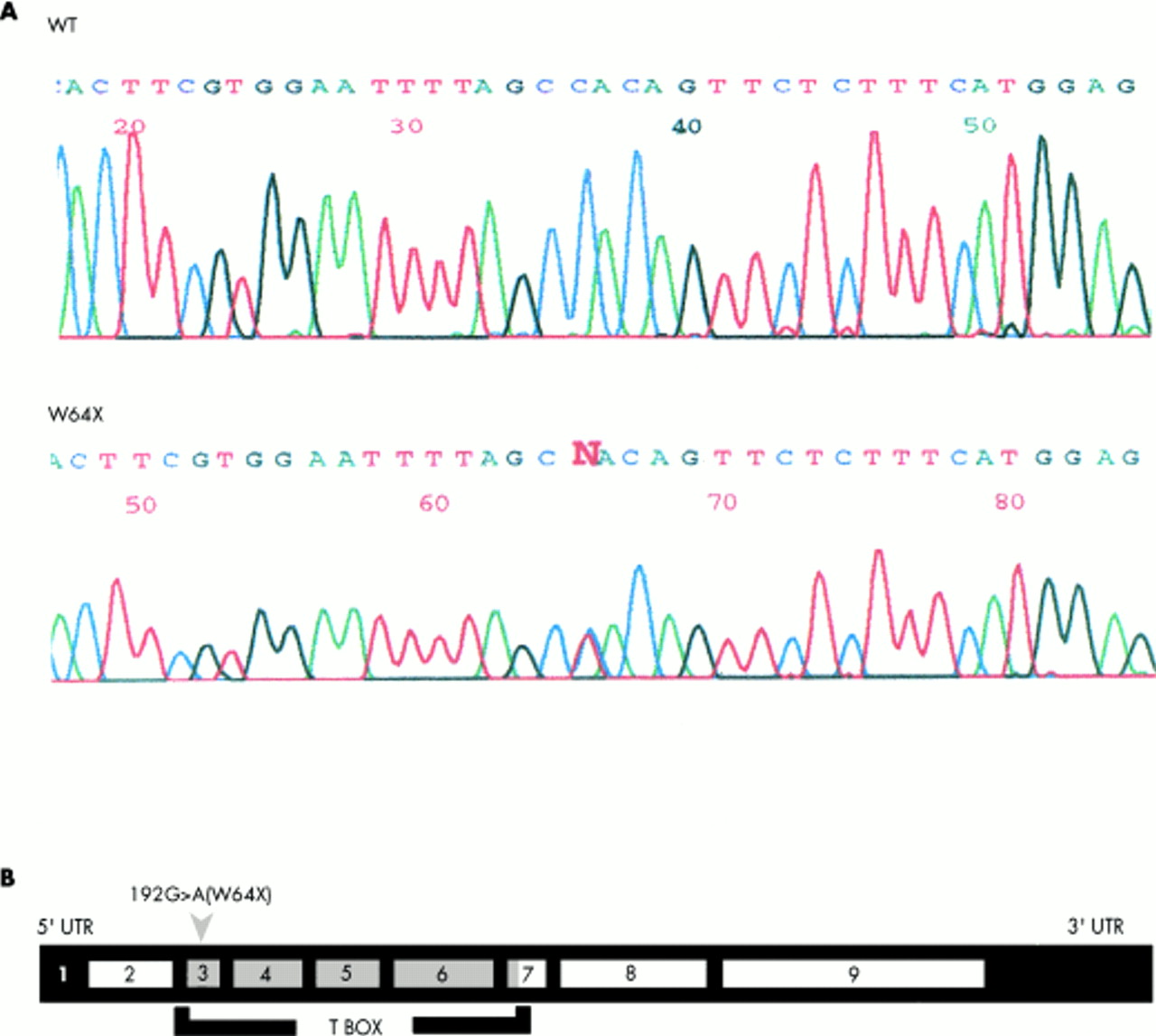
Identification of a novel TBX5 nonsense mutation, 192G>A(W64X), associated with a single Holt-Oram syndrome patient (HOS002). (A) Sequence analysis of TBX5 exon 3 for a normal subject (WT) and the single Holt-Oram syndrome patient (HOS002). The conspicuous “N” in panel HOS002 indicates a heterozygous state, indication of the presence of the WT allele (C) and the mutant allele (t). The sequences are antisense. (B) The substitution of c by t (g by a on the complementary strand) leads to the change of codon 64 of TBX5 coding for tryptophan into a stop codon, which truncates TBX5 polypeptide by 88%. The 192G>A(W64X) mutation is located in exon 3 of TBX5.
The DNA binding activity of TBX5 is altered by the 381-408del27bp deletion
Because the 381-408del27bp deletion results in an in frame deletion of only nine amino acids in TBX5, its functional significance is not known. Thus, we assessed the ability of the mutant TBX5 protein to bind to the target DNA binding site using gel shift experiments. The upstream region of the promoter of ANF (the atrial natriuretic factor gene) contains a TBX5 binding site.18 The TBX5 binding site was chemically synthesised as pairs of complementary single strands (fig 3). The synthetic site was labelled and used in gel retardation experiments with in vitro translated TBX5 proteins. As shown in fig 3, the wild type TBX5 protein forms a DNA protein complex. Competition experiments with unlabelled synthetic TBX5 binding site and a synthetic fragment with a sequence unrelated to the TBX5 binding site suggested that the observed binding of TBX5 to the synthetic binding site is specific (data not shown). In contrast, in the lane with the TBX5 protein with the 381-408del27bp deletion, we observed a smaller and very weak band, which may represent the complex between the mutant TBX5 and DNA (fig 3). The intensity of TBX5-DNA complex in the WT lane was estimated to be 5.8-fold stronger than that in the 27 bp deletion lane. These data indicate that mutant TBX5 protein with the 381-408del27bp deletion greatly reduces the DNA binding activity of TBX5. Next, we tested whether the binding capability of the wild type TBX5 to DNA could be reduced by addition of the mutant TBX5 protein. As shown in fig 3, the 381-408del27bp deletion did not interfere with the binding of wild type TBX5 to DNA.
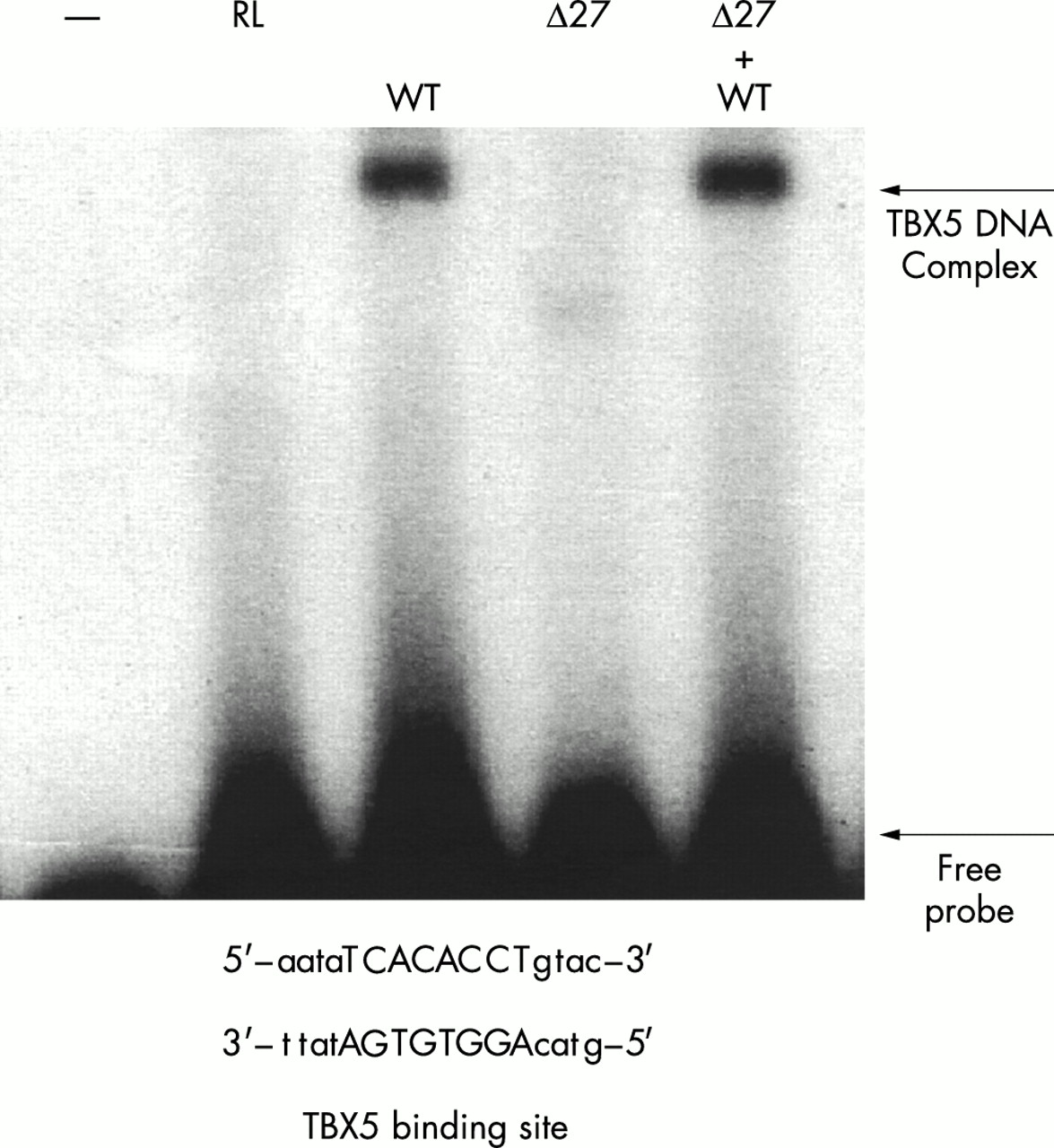
Gel retardation analysis of TBX5 binding to DNA. Lane indicated by symbol −, free DNA; lane RL, rabbit reticulocyte lysate or TNT T7 Quick Master Mix; lane WT, wild type TBX5; Δ27, mutant TBX5 containing the 381-408del27bp deletion; lane Δ27+WT, mixture of wild type TBX5 and mutant TBX5 with the 381-408del27bp deletion.
The ability of TBX5 to activate the ANF promoter is altered by the 381-408del27bp deletion
The functional effect of the TBX5 381-408del27bp mutation on transcription was assessed using a reporter gene (ANF-luc) with the ANF promoter (region from –270 to + 1) fused to the luciferase gene.18 The ANF-luc construct was cotransfected with various TBX5 expression constructs in HEK-293 cells. Western blot analysis showed that the mutant TBX5 with the 381-408del27bp deletion was stably synthesised by transfected cells (fig 4A). The transcriptional activity was examined and expressed as relative luciferase units. As shown in fig 4B, over-expression of the wild type TBX5 activated transcription from the ANF promoter. The 381-408del27bp deletion completely abolished the transcription of the ANF promoter (fig 4B). Coexpression of mutant TBX5 with wild type TBX5 showed the similar transcriptional activities to wild type TBX5 alone. Together, these data suggest that the 381-408del27bp deletion causes dysfunction in transcriptional activation of TBX5.

The 381-408del27bp deletion dramatically reduces transcriptional activation of the ANF promoter. (A) Western blot analysis of proteins from HEK-293 cells expressing FLAG tagged wild type (WT) and mutant TBX5 with 381-408del27bp deletion. The positions of molecular weight markers are indicated. The mouse anti-FLAG M2 primary antibody detected a band with the expected size of TBX5 (∼58 kDa). (B) The ANF-luciferase reporter gene was cotransfected with different TBX5 expression constructs in HEK-293 cells, and the level of transcriptional activation was expressed as relative luciferase activity. Vector, pcDNA3.1; WT, wild type TBX5 expression construct; Δ27bp, mutant TBX5 expression construct containing the 381-408del27bp deletion; Δ27bp+WT, cotransfection of wild type and mutant TBX5 expression constructs.
Nuclear localisation of TBX5 is impaired by the 381-408del27bp deletion
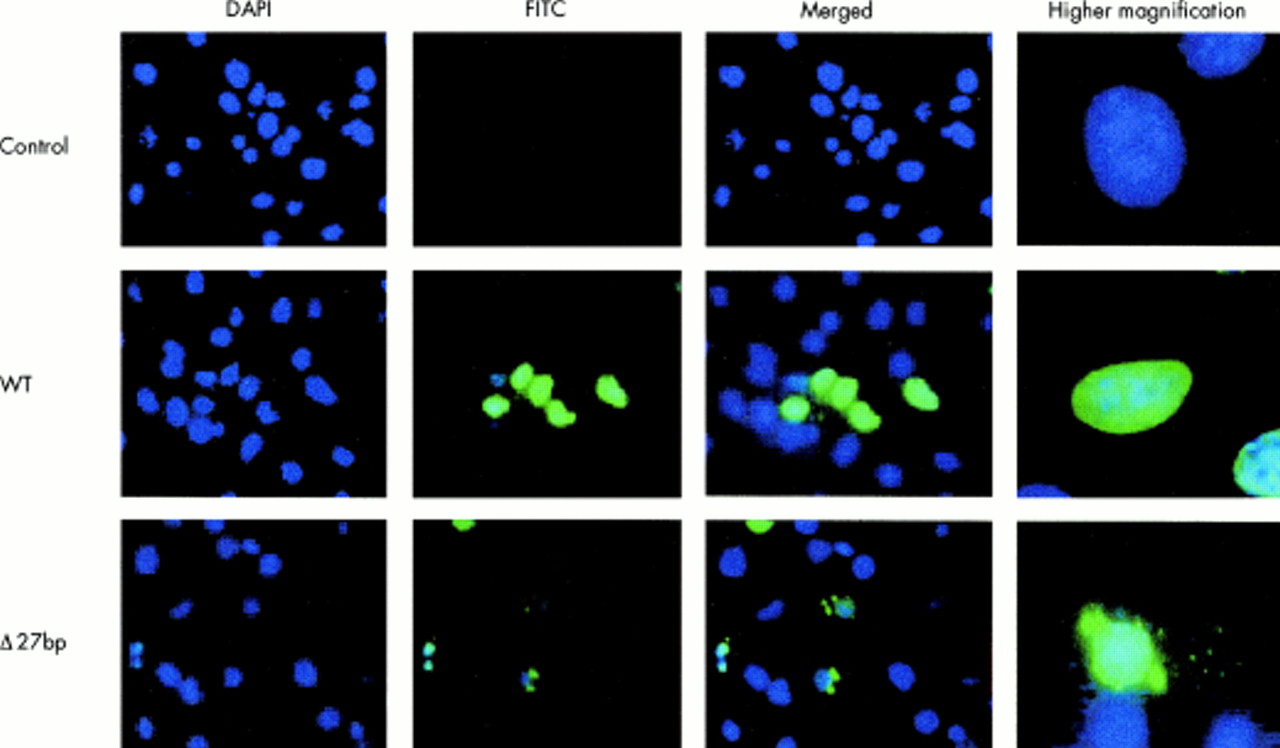
To characterise cellular localisation of wild type and mutant TBX5 proteins, NIH-3T3 cells were transfected with FLAG tagged TBX5 expression constructs. Immunofluorescence staining showed that, as expected, wild type TBX5 was localised to nuclei (fig 5, WT, green signal). However, nuclear localisation of TBX5 was altered in cells transfected with the 381-408del27bp deletion mutant. Instead, immunofluorescence signal was found in both nuclei and cytoplasm, with prominent staining as aggregates in the cytoplasm (fig 5, Δ27bp). Similar results were obtained with HEK-293 cells (data not shown). These results suggest that the 381-408del27bp deletion impairs nuclear localisation of TBX5.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immunostaining of NIH 3T3 cells expressing the FLAG tagged wild type (WT) and mutant 381-408del27bp (Δ27bp) TBX5. Control, cells transfected with vector pcDNA3.1. Transfected cells were immunostained with anti-FLAG (FITC, green) for TBX5 and with DAPI for nuclei (blue). Merged, combination of DAPI and FITC. Images of single cells at a higher magnification are shown in the right column. Similar results were obtained with HEK 293 cells (data not show).
DISCUSSION
Here we report the identification and characterisation of a novel 381-408del27bp deletion in the TBX5 gene in a large family affected with Holt-Oram syndrome (HOS), and a novel 192G>A(W64X) nonsense mutation in a single Holt-Oram syndrome patient. The 381-408del27bp deletion and the 192G>A(W64X) mutation identified in this study expand the spectrum of TBX5 mutations causing Holt-Oram syndrome.20
TBX5 haploinsufficiency (loss of an expressed allele) has been hypothesised as a mechanism for Holt-Oram syndrome. This hypothesis is supported by identification of translocation t(5;12) in intron 1 of TBX5 that separates protein coding exons 2-9 from the promoter and 5′-UTR and the recent study showing that Tbx5del/+ mice modelled Holt-Oram syndrome.17,22 Nonsense mediated mRNA decay is another mechanism that can lead to haploinsufficiency.28 Thus, the molecular mechanism by which mutation 192G>A(W64X) causes Holt-Oram syndrome may be nonsense mediated decay.
The net effect of haploinsufficiency is 50% reduction of TBX5 in the patients with Holt-Oram syndrome. The similar effect is expected for the 381-408del27bp deletion in TBX5 identified in this study. Western blot and immunostaining studies clearly showed that TBX5 with the 381-408del27bp deletion was stably expressed in two different types of cells, NIH3T3 and HEK-293 cells (fig 4A, fig 5). However, the 381-408del27bp deletion dramatically reduced the capabilities of TBX5 to bind to DNA and to activate transcription. Our DNA binding and transcription activation analyses showed that the existence of the mutant TBX5 in the context of wild type TBX5 did not affect the function of wild type TBX5 (figs 3 and 4). In vitro, mutation 192G>A(W64X) also eliminated DNA binding and transcription activation activities of TBX5, and did not affect the function of wild type TBX5 (data not shown). These data exclude a dominant negative effect as the molecular mechanism for Holt-Oram syndrome.
Immunofluorescence staining of cells (NIH3T3, HEK-293) transfected with tagged TBX5 proteins showed that wild type TBX5 was localised in the nucleus, but some mutant TBX5 molecules with the 381-408del27bp deletion were retained in the cytoplasm (fig 5). These results show impaired nuclear localisation of TBX5 as a novel mechanism for Holt-Oram syndrome. The mechanism of the retention of mutant TBX5 in the cytoplasm is not clear. It is likely that the 381-408del27bp deletion results in misfolded protein that impairs TBX5 transport and trafficking. Some partly folded or incorrectly folded mutant TBX5 may generate aggregates of varying size (fig 5), which may have difficulty entering the nucleus. The misfolding induced aggregates do not have function and are associated with some human disease.29 TBX5 does not contain an obvious nuclear localisation signal, thus its nuclear localisation domain (NLD) is unknown. The 381-408del27bp deletion of TBX5 may also affect the function of NLD, resulting in retention of some mutant molecules in the cytoplasm. It is interesting to note that trafficking defects caused by disease related mutations may appear to be a general rule for cardiovascular disease, because in addition to Holt-Oram syndrome studied here such defects have been reported for long QT syndrome30 and SCN5A mutations causing Brugada syndrome.31
Newbury-Ecob et al3 analysed 55 cases with Holt-Oram syndrome from the United Kingdom, and found that the phenotype of Holt-Oram syndrome displayed increasing severity in succeeding generations. In the large three generation Holt-Oram syndrome family presented in this study, we also observed increasing severity of the cardiac manifestation in successive generations. The affected male in the first generation (fig 1) has skeletal defects only and exhibits no cardiac defects. The five affected females in the second generation exhibit no major cardiac defects except mitral valve prolapse (MVP) and incompetence (fig 1). MVP can occur in 4-8% of young adults and more often in females than in males,32 so it is not clear whether MVP observed in the family is related to the 381-408del27bp deletion of TBX5. In contrast, eight patients in the third generation had atrial septal defects and ventricular septal defects that required early surgery, and one patient died from multiple cardiac defects (fig 1). Using the severity score system that was developed for Holt-Oram syndrome by Newbury-Ecob et al,3 we estimated the average severity scores of the cardiac abnormalities for generations I, II, and III to be 0, 0.6, and 1.85, respectively, which are significantly different (p=0.05). The increasing severity of cardiac defects in succeeding generations of Holt-Oram syndrome families is consistent with the phenomenon of anticipation. Anticipation is defined as increasing severity/incidence and/or decreasing age at onset in successive generations, and it is caused by dynamic mutations that are expanding, unstable DNA repeat sequences such as the trinucleotides CCG/CGG and CAG/CTG.33 However, it is important to note that the phenomenon of anticipation usually involves decreased age of onset in the succeeding generations, which is not applicable to a congenital heart defect. Furthermore, the finding of possible anticipation in the Holt-Oram syndrome families in this and other studies may simply represent ascertainment bias, reproduction selection,3 or a stochastic chance.
In conclusion, we have identified two novel TBX5 mutations, 381-408del27bp (the 27 bp in frame deletion) and 192G>A(W64X) (a nonsense mutation), which are associated with Holt-Oram syndrome. Functional analysis of the 381-408del27bp deletion suggests that impaired nuclear localisation as well as dysfunction in DNA binding and transcriptional activation are two distinct mechanisms that can account for the loss of functional TBX5 proteins in Holt-Oram syndrome.
Acknowledgments
The first two authors contributed equally to this work. We thank Shaoqi Rao for statistical analysis, Gaston Magri and Yolanda Farre for patient care, and Hilda Novo for technical assistance. This study was supported by a Fourjay Foundation Cardiovascular Research Grant (QW) and NIH grants R01 HL65630 and R01 HL66251 (QW).