Article Text

Statistics from Altmetric.com
The neurofibromatoses are a group of neurocutaneous disorders that show extreme clinical heterogeneity and are characterised by growth abnormalities in tissues derived from the embryonic neural crest.1,2 Two main clinical forms exist, type 1 (NF1) and type 2 (NF2), as well as several alternate and related forms.2,3 NF1 and NF2 are the only clinically well defined disorders and both genes have been identified.4–8 The NIH diagnostic criteria for NF1, as defined by the conference statement,9 are met if two or more of the following are found: six or more CAL spots; two or more neurofibromas of any type or one plexiform neurofibroma; axillary or inguinal freckling; optic glioma; two or more Lisch nodules; a distinct osseous lesion; a first degree relative (parent, sib, or offspring) with NF1 according to the above criteria.
Spinal nerve sheath tumours are described as symptomatic findings in only 5% of NF1 patients,10 although they can be observed by MRI in up to 36% of patients.11–13 The presence of a wide, symmetrical distribution of spinal neurofibromas, occurring in all adult affected members of the same family and segregating in an autosomal dominant fashion, is however extremely rare. This form, familial spinal NF (FSNF), has been considered an alternate form of NF since patients generally lack dermal neurofibromas and Lisch nodules, both typical hallmarks of NF1, and since symptomatic and generalised spinal neurofibromas are uncommon in classical NF1. FSNF has been reported in only four families.12,14,15 Three multigenerational families with spinal neurofibromas and CAL spots were shown to be linked to markers surrounding the NF1 locus.12,14,15 In the fourth family, presenting with spinal neurofibromas without CAL spots, linkage to the NF1 locus was excluded.14 Only in one FSNF family has the underlying molecular defect been documented so far,15 which was a unique frameshift mutation 8042insA in NF1 exon 46. Here we describe the identification of the NF1 mutation in both remaining FSNF families originally described by Pulst et al14 and Pöyhönen et al.12 Our current findings emphasise that FSNF (with CAL macules) is caused by mutations in the NF1 gene, but does not support the hypothesis that it is caused by a unique type of NF1 mutation.
MATERIALS AND METHODS
Subjects
The pedigrees of the families studied are shown in figs 1A and 2.
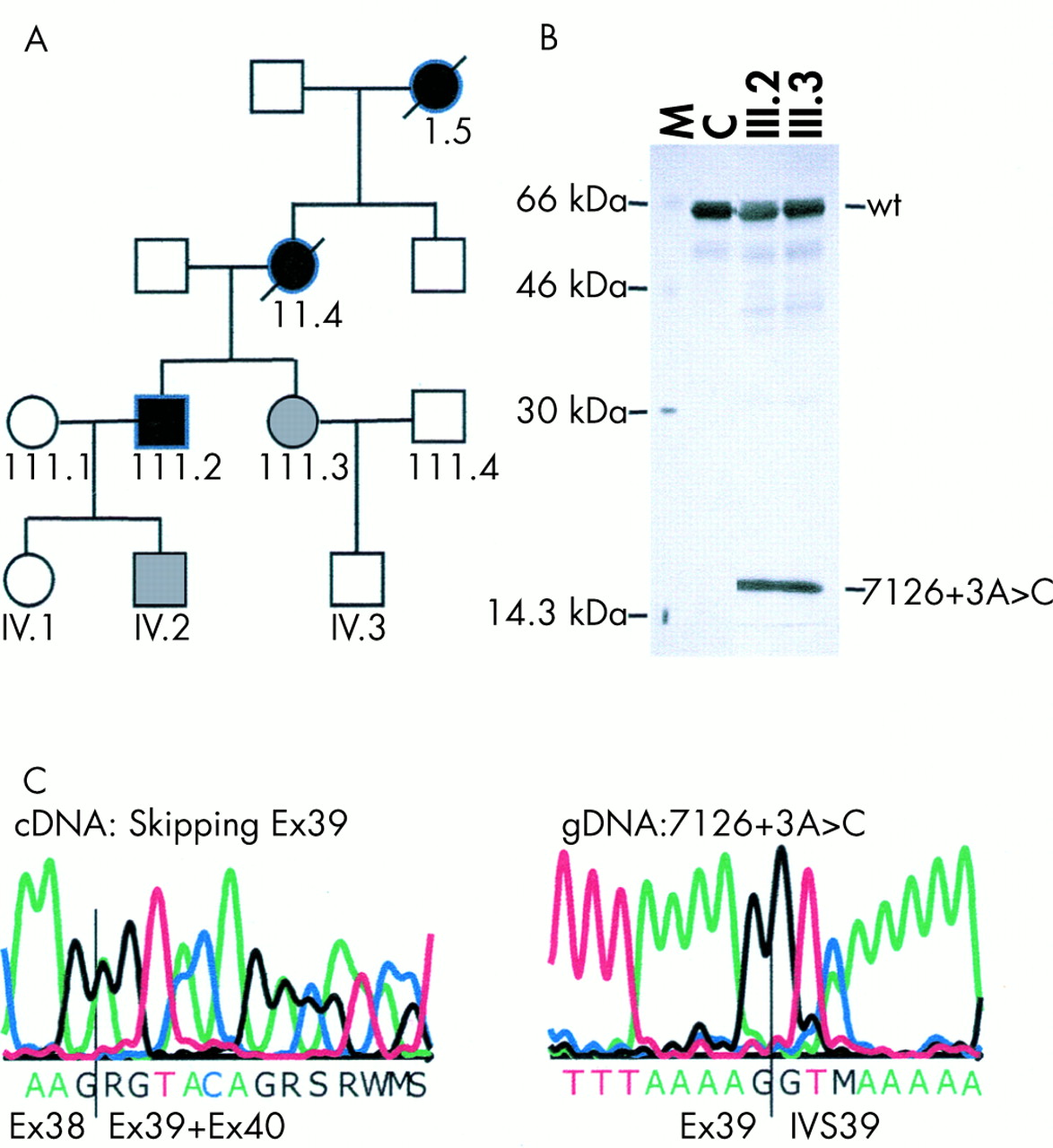
Pedigree and PTT results of family 1 with mutation 7126+3A>C. (A) In the pedigree, blackened symbols denote affected subjects with spinal neurofibromas and CAL spots, grey symbols denote affected subjects with CAL spots only, and white symbols denote healthy subjects. (B) PTT results using primers encompassing exons 34 to 49 of a control (C) and of patients III.2 and III.3. The wild type (wt) and aberrant band caused by skipping of exon 39 are shown. M denotes a protein marker with sizes in kDa. (C) Direct cycle sequencing of mutant cDNA transcripts and genomic DNA. By direct cDNA sequencing of the patient, heterozygosity for transcripts containing exon 39 and transcripts in which exon 39 is skipped are seen. In the genomic DNA, heterozygosity for A and C at position +3 of the splice donor site of exon 39 is seen in the patient.

Pedigree of family 2 with mutation L357P. In the pedigree, blackened symbols denote affected subjects with spinal neurofibromas and CAL spots, grey symbols denote affected subjects with CAL spots only, and white symbols denote healthy subjects.
Skin samples of controls were obtained from operations carried out for cosmetic reasons from four healthy persons at the Department of Dermatology, University of Oulu, Finland, with the consent of the Ethical Committee of Oulu University Hospital.
NF1 mutation analysis
Epstein-Barr virus (EBV) lymphoblastoid cell cultures from two affected members of family 1 and fibroblast cell cultures from two affected members of family 2 were treated with and without puromycin before total RNA extraction as described previously.16 DNA was extracted from EBV cell cultures or fibroblasts from all family members.
Key points
-
Familial spinal neurofibromatosis (FSNF) is considered to be an alternate form of neurofibromatosis, with a limited phenotype of multiple spinal nerve root neurofibromas and café au lait (CAL) macules in all affected adults. Only three multigeneration families with FSNF have been reported and in one of them the genetic defect was identified previously: 8042insA in exon 46 of the NF1 gene.
-
As NF1 is notorious for its extreme phenotypic variability even within the same family, the striking homogeneity of the phenotype in these families suggests that a particular type of mutation might underlie this condition.
-
We identified the NF1 mutation in the remaining two FSNF families whose phenotype was known to be linked to chromosome 17q11.2 markers. In the first family, a novel substitution at the splice donor site of exon 39 (7126+3A>C), leading to skipping of exon 39, was found. In the second family, a missense mutation L357P in exon 8 was identified as the sole alteration. The mutation segregates with the disease in both families.
-
The current findings emphasise that FSNF (with CAL macules) is caused by mutations in the NF1 gene, but does not support the hypothesis that it is caused by a specific type of NF1 mutation. It is anticipated that expression studies and mutation analysis, in the first instance of the genes tightly linked to the NF1 locus, in both families may lead to the detection of (a) modifier(s) for this specific phenotype.
The optimised PTT for the entire coding region was applied essentially as described previously with abnormal fragments further analysed by cDNA and genomic sequencing.16 Then, for both families, the entire NF1 cDNA was sequenced using dye-primer chemistry on an automatic genetic analyser (ALFexpress). Information on the sequencing primers used is available upon request.
Western blot analysis
Western blot analysis, using an anti-NF1 antibody (NF1GRD(D)) (Santa Cruz Biotechnology Inc, Santa Cruz, CA) and peroxidase linked donkey anti-rabbit (NA 934) (Amersham International plc, Little Chalfont, Buckinghamshire, England) as a secondary antibody, was carried out essentially as previously described.17
RESULTS
Clinical evaluation
In both families all affected adults, except for one 45 year old female in family 1 (III.3) showed multiple symmetrically distributed spinal neurofibromas in the cervical, thoracic, and/or lumbar region (fig 3). All 12 affected members in both families also had more than six (size over 15 mm) cutaneous CAL spots, but no iris Lisch nodules were present. Clinical data, originally described by Pulst et al14 and Pöyhönen et al,12 have been updated and are summarised in table 1.
Clinical features of affected patients in FSNF families 1 and 2

{kind=link}
{kind=link}
{kind=link}
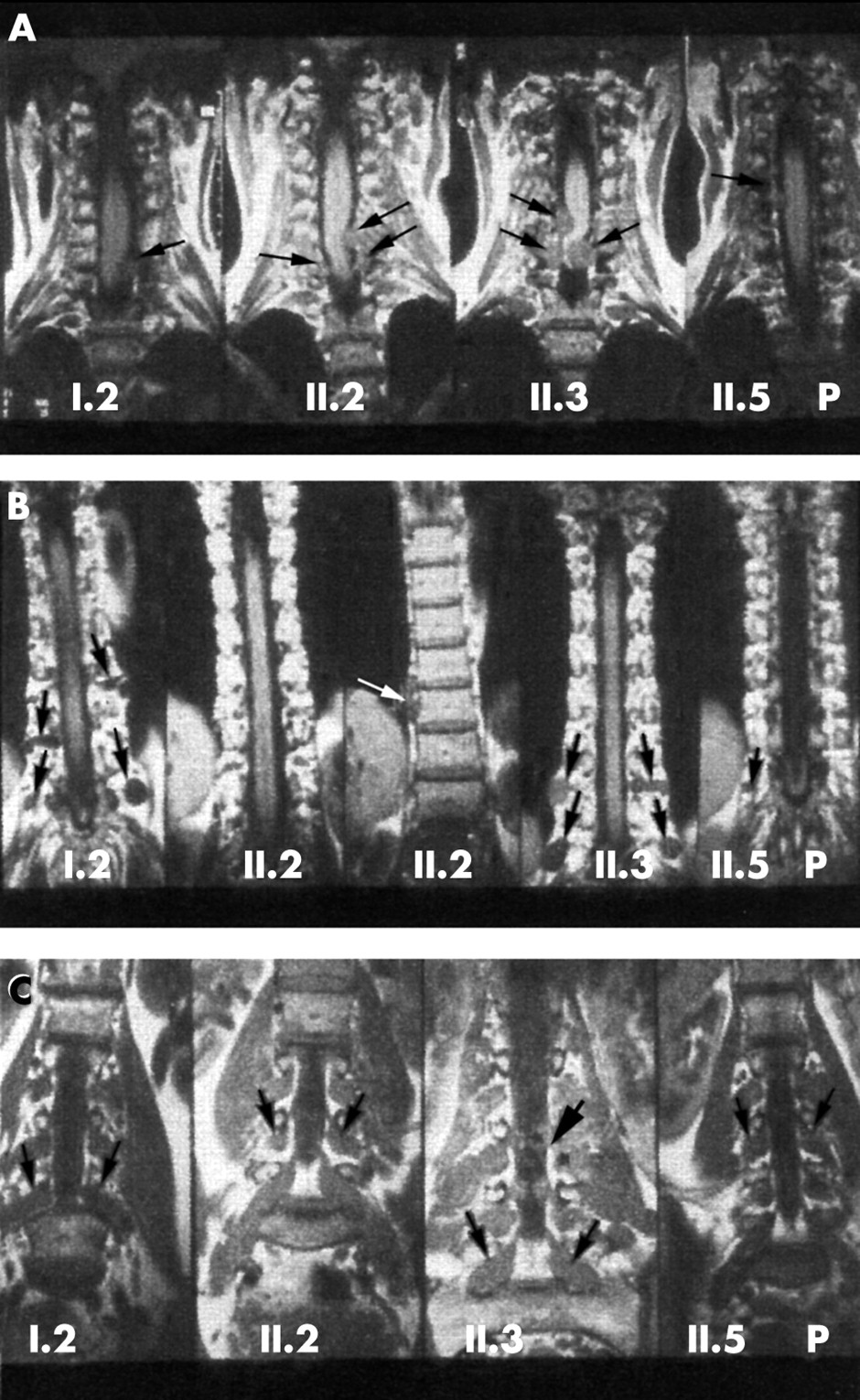
Coronal T1 weighted non-contrast MR images (Siemens Magnetom 1.0T, SE, TR 500, TE 15, slice thickness 3 mm) of patients I.2, II.2, II.3, and II.5 (P=proband) from family 2, carrying the mutation L357P. (A) Cervical spine. (B) Thoracic spine. (C) Lumbar spine. Several extradural, intraspinal neurofibromas compressing the spinal cord (black curved arrows) and extraspinal neurofibromas (black straight arrows) are seen in all patients. Not all tumours are marked. II.2 also has a right sided paraspinal thoracic mass (white arrow). The intra- and extraspinal components of a dumb-bell tumour in the lumbar spine (thick black arrow) in patient II.3 are shown.
Molecular analysis
In family 1, PTT analysis of the NF1 cDNA of patients III.2 and III.3, encompassing exons 35 to 49, showed in addition to the wild type 68.5 kDa product a truncated polypeptide of approximately 19 kDa, suggesting a truncating mutation between nucleotides 6900 and 7200 (fig 1B). Sequencing of the RT-PCR product showed absence of the whole of exon 39 (fig 1C). By genomic DNA sequencing, a novel substitution, 7126+3A>C (fig 1C), was found at the splice donor site of exon 39. This is a novel mutation not identified previously in classical NF1 patients. We calculated the splice site donor strength from the wild type sequence (7126+3A) and the mutant sequence (7126+3C) using the algorithms developed by Shapiro and Senepathy18 (S&S) and using the Splice Site Prediction by Neural Network (SSPNN, http://www.fruitfly.org/seq_tools/splice.html) program. The scores for the mutant sequence (69.9 (S&S) and 0.09 (SSPNN)) were significantly lower than the wild type sequence scores (79.9 (S&S) and 0.82 (SSPNN)). Hence, less efficient binding of splice factors at the mutant splice donor site will modulate the splicing efficiency leading to skipping of exon 39. In order to exclude the presence of a putative second alteration in the NF1 gene, the complete NF1 cDNA was sequenced using direct cycle sequencing, but no other pathogenic alteration was found. 7126+3A>C was present in all affected subjects (II.4, III.2, III.3, and IV.2) and was absent in the healthy family members (III.1, III.4, IV.1, IV.3).
In family 2, only normal sized fragments were discerned by PTT analysis of the total coding region, indicating the absence of a truncating mutation. Later, direct cycle sequencing of the total NF1 cDNA was performed and a missense mutation was identified in exon 8 as the sole alteration, changing leucine at amino acid 357 to proline (L357P). Patients I.2, II.2, II.3, II.5, III.1, III.3, and III.4 were shown to bear the mutation L357P, whereas the healthy family members II.1 and III.2 did not carry this mutation. This indicates that the mutation segregates with the disease. Furthermore, this alteration was not found by analysing 200 normal chromosomes. L357 is also conserved in Fugu rubripes (AF064564), Drosophila (L26501), mouse (L10370), and rat (D45201). Proline is a very rigid amino acid and its presence creates a fixed kink in a polypeptide chain, which might have dramatic consequences for the protein structure. Altogether, these data indicate that L357P is the pathogenic NF1 mutation in this family.
Western blot analysis
Four fibroblast cell cultures from normal subjects and three fibroblast cell cultures initiated independently from the normal skin of patient II.5 carrying the missense mutation L357P (family 2) were analysed by Western transfer analysis. This showed that the levels of NF1 protein were slightly higher in three out of four fibroblast cell lines from normal subjects compared to fibroblasts cultured from patient II.5 (data not shown).
DISCUSSION
Familial spinal NF has been considered an alternate form of NF1 because of the exceptional occurrence of multiple spinal neurofibromas affecting spinal roots symmetrically in all affected adult members of the same family accompanied by very mild cutaneous signs of NF1 and absence of Lisch nodules.3 Only three such well documented families have been described and CAL spots were considered the sole cutaneous sign present in all affected members.12,14,15 Although the clinical manifestations in the family members fulfil the NIH criteria for NF1 (presence of more than six CAL spots, two neurofibromas of any type, in this case multiple spinal neurofibromas, and also a first degree relative (parent, sib, or offspring) with NF1), they are very atypical of NF1. First, freckles, dermal neurofibromas, and Lisch nodules are found in more than 80%, 90%, and 90% respectively of classical NF1 patients but these signs were generally absent in all affected members of the FSNF families studied. One affected member had freckles in the axillary area and two had dermal neurofibromas, but no Lisch nodules were seen. Secondly, symptomatic spinal neurofibromas are rare in NF1 patients (<5%). Furthermore, the presence of multiple spinal neurofibromas affecting all spinal roots symmetrically in affected patients leading to severe neurological impairment is generally atypical of NF1, a disease noted for its variable expressivity among family members with identical NF1 mutations. These very specific clinical findings suggest that a genotype-phenotype correlation might be found in these families and that a specific type of gene defect with a special effect on neural crest cells and/or their precursors in the nerve roots might be present in these families.
In one of these families, the pathogenic lesion had been identified previously, a unique frameshift at the 3‘ end of the NF1 gene in exon 46, 8042insA.15
We have now identified a bona fide pathogenic mutation in the affected members of both the other families. In family 1, a splice donor mutation was identified (7126+3A>C) resulting in out of frame skipping of exon 39 at the mRNA level. This is a novel mutation not identified previously in classical NF1 patients. In family 2, a recurrent missense mutation in exon 8 was found (L357P). This mutation was reported previously19 in an NF1 patient, but no clinical findings are available so we are unable to correlate this genotype with the specific phenotype.
The current findings underline that FSNF with CAL spots is not only linked to the region flanked by the markers HHH202 and pEW206, encompassing the NF1 locus, but is clearly allelic with NF1. Furthermore, taking the data from Ars et al15 and ours together, it is clear that all three well defined FSNF families carry a different private NF1 mutation, as also is the case for classical NF1. Moreover, it is unlikely that a specific type of mutation located in a specific region of the gene underlies this phenotype, as a frameshift, a splice mutation, and a missense mutation were found, all located in different parts of the gene. However, it is noteworthy that the mutations found in the three FSNF families might be mild mutations with some residual function as they are a truncating mutation at the very 3‘ end of the gene 8042insA, a splicing error 7126+3A>C, and a missense mutation L357P. Recently, Kaufmann et al20 described two families with multiple spinal tumours without CAL spots. Although overall the cutaneous signs were very mild in the affected patients, several of them had cutaneous neurofibromas. In these families too, the underlying NF1 mutation was a splicing (IVS31-5A>G) and a missense mutation (L2067P). It has been proposed that the FSNF phenotype might arise through a negative residual function of the mutant neurofibromin with a special effect on the development of the neural crest cell.15 When a total gene deletion or a nonsense mutation in the gene is encountered, these mutations will result in a null allele or in transcripts that become degraded by the nonsense mediated RNA decay, leading to haploinsufficiency. For the mutations found in the FSNF families, however, it is conceivable that a negative residual function might still exist, that is, that a mutant neurofibromin with the single amino acid change is formed by the L357P allele, a 2680 amino acid truncated protein is formed by mutation 8042insA, and a 2353 amino acid truncated protein is formed by 7126+3A>C, albeit probably in a minimal amount. This assumption was tested by western blotting of the EBV cell lines of two patients carrying 7126+3A>C, using the antibody SC67, but no shortened neurofibromin was found (data not shown). However, we do not think that this is enough evidence to draw definitive conclusions as the molecular tools available for these studies are still limited today.
Mild mutation can however not be the sole factor predisposing to the development of multiple spinal tumours, as a number of similar frameshift mutations at the 3‘ end of the NF1 gene, splicing, and missense mutations were found in patients without spinal involvement16,19,21 (unpublished data) and with classical cutaneous manifestations. It is generally accepted that bona fide missense mutations may point to critical functional domains in a protein19 as they may lead to the production of a mutant protein. The RasGAP activity of the central GAP related domain as well as the structure of the GRD of neurofibromin have been well characterised22,23 and the effects of specific missense mutations in this domain have been studied in detail.24 No functional significance has been contributed so far to the region encompassing exon 8 in which the L357P mutation was found. Recently, the neurofibromin content of cells from three NF1 patients with spinal neurofibromas without CAL spots was found to be reduced to half of the amount of normal control cells, suggesting functional haploinsufficiency.20 Interestingly, one of the mutations tested was a missense mutation L2067P. In order to substantiate these findings further, we have performed western blot analysis on control normal fibroblast cell cultures (n=4) and fibroblast cell cultures (n=3), independently initiated from patient II.5, carrying the mutation L357P. The levels of NF1 protein were slightly higher in three out of four control normal fibroblast cell lines compared to the L357P cell lines. No apparent differences were noted between fibroblasts from one normal control compared to patient II.5 with FSNF. This most probably is because the NF1 gene is not a housekeeping gene and the same cell line can display marked expression changes when analysed at different times.25 However, we do not think that this evidence is enough to form a definitive conclusion of haploinsufficiency. It was also proposed that the FSNF was the result of the presence of a second NF1 mutation, either on the normal allele (in trans) or, more likely, on the same allele that also contains the already identified mutation (in cis).15 As we applied the complete cascade of techniques used to find the mutation in >95% of classical NF1 patients16 and did not find a second alteration, it is unlikely that a second change in the NF1 gene is present concomitantly.
Finally, it is possible that an additional mutation in another gene lying within the region flanked by the markers HHH202 and EW206 and shown to be linked in both families influences the striking clinically homogeneous phenotypic outcome. Analysis of additional highly polymorphic microsatellite markers in both families will allow narrowing down of the linked region of interest for further investigation. It has been suggested that a modifier gene resides in close proximity to the NF1 gene26 and that its deletion together with the NF1 gene results in the severe “NF1 microdeletion” phenotype with early onset of growth and an excessive number of cutaneous neurofibromas. A mutation (for example, with a dominant negative effect on the development of the neural crest cells that are responsible for the NF1/FSNF phenotype) in one of the genes flanking the mutant NF1 gene might cooperate with the NF1 mutation to result in FSNF. Furthermore, the same type of alteration, present in trans with an NF1 mutation or occurring as a somatic event may be the modifier causing spinal tumours in a proportion of NF1 patients, but not in their offspring carrying only the NF1 mutation. Expression and mutation analysis of the genes residing in this region, in the patients from FSNF families and in isolated patients presenting with this specific phenotype, will shed more light on this hypothesis and should help in understanding the molecular alterations that cause this severe neurological phenotype.
Acknowledgments
This work was supported by a grant from Ghent University to IV (BOF 01107799 and 011D3801) and by a grant from the Interuniversity Attraction Poles P5 to LM. This work was supported by grants DAMD17-00-1-0542 and DAMD 17-01-1-0721 awarded to KS and by grants from the Finnish Cancer Societies (JP) and Oulu University Hospital (JP). We wish to thank the patients and their families for excellent cooperation.