Article Text

Statistics from Altmetric.com
- VHL, von Hippel-Lindau disease
- cHAB, central nervous system haemangioblastoma
- rHAB, retinal haemangioblastoma
- RCC, renal cell carcinoma
- ELST, endolymphatic sac tumour
- PNET, pancreatic neuroendocrine tumours
- APMO, adnexal papillary tumour of probable mesonephric origin
- QSA, quantitative Southern analysis
- HIF, hypoxia inducible factor
- UPQFM-PCR, universal primer quantitative fluorescent multiplex polymerase chain reaction
- LOH, loss of heterozygosity
Von Hippel-Lindau (VHL) disease is a rare autosomal dominant disorder characterised by a predisposition to haemangioblastomas of the central nervous system (cHAB) and retina (rHAB), renal cell carcinomas (RCC), phaeochromocytomas and paragangliomas, endolymphatic sac tumours (ELST), pancreatic neuroendocrine tumours (PNET), papillary cystadenomas of epididymis, and adnexal papillary tumours of probable mesonephric origin (APMO). Renal, pancreatic, epididymal, and broad ligament cysts also occur frequently.1 It is caused by germline mutations in the VHL tumour suppressor gene on chromosome 3p25-26.2,3 VHL disease occurs with an estimated incidence of 1:36 000 live births and shows variable expression and age dependent penetrance that is almost complete by the age of 65.4 Median actuarial life expectancy of VHL subjects is reduced to 49 years, renal cell carcinoma5 or CNS haemangioblastoma6 being the most common cause of death. At present, early diagnosis and treatment of VHL associated tumours result in improved prognosis for VHL subjects.7
The disease can be diagnosed on the basis of clinical criteria in the presence of a single haemangioblastoma, phaeochromocytoma, multiple pancreatic cysts, or renal cell carcinoma in a member of a VHL family. For clinical diagnosis of isolated cases of VHL, at least two haemangioblastomas or a single haemangioblastoma in association with a visceral manifestation are required.8 In early studies, using standard Southern analysis and sequencing of the VHL coding region, disease causing mutations were detected in 38-80% of families fulfilling clinical VHL criteria.1 More recently, the development of quantitative Southern analysis (QSA) to detect partial and complete deletions has improved VHL mutation detection. QSA analysis in conjunction with direct sequencing of the coding region showed germline mutations in 100% of 93 families with definite VHL.9 However, the results implying that all cases fulfilling clinical VHL criteria are related to mutations in the VHL gene should be verified by other studies, and by the use of recently developed mutation detection strategies.
The spectrum of constitutional mutations in the VHL gene includes changes such as single base substitutions, microdeletions, and microinsertions identified in ∼2/3 of affected families and large deletions observed in ∼1/3 of VHL families. Large deletions involve a part of or the entire VHL gene. A few common VHL germline mutations have been described (233A>G, 226-228delTTC, 481C>T, 499C>T, 500G>A, 533T>C).9–18 In most cases these have multiple origins and represent a high de novo mutation rate in hypermutable sequences.19 A founder effect was reported only for the 292C>T substitution in families from Germany.20 Germline de novo mutations may be present in as many as 23% of VHL patients, since first generation diagnosis has been reported in 42 of 181 VHL kindreds evaluated by NIH.21
The VHL gene product (pVHL) has two macromolecular sites, α and β domains, which can bind different proteins. pVHL bound to elongin B, C/cullin/Rbx1 acts as an E3 ubiquitin ligase and targets proteins for degradation.22 Similarly, hypoxia inducible proteins are regulated by complex pVHL/elongin B and C that requires direct binding of hypoxia inducible factor (HIF) to the β domain and targets HIF for ubiquitination catalysed by the α domain.23 The VHL protein also plays a role in extracellular matrix formation and is necessary for cell cycle exit.24,25 Germline VHL mutations cause different protein defects that result in the clinical heterogeneity of VHL disease. VHL type 1 disease (without phaeochromocytoma) is associated with mutations predicted to cause complete unravelling of protein structure (missense mutations mapping to VHL protein hydrophobic core, protein truncating mutations, and partial gene deletions). VHL type 2 (with phaeochromocytoma) is caused by missense mutations which map to protein binding sites of pVHL and are predicted to cause local protein defects.22 Specific phaeochromocytoma associated missense mutations confer different risks of HAB and RCC, being responsible for the occurrence of subcategories VHL type 2A (VHL without RCC), 2B (VHL with RCC), and 2C (phaeochromocytoma only).15 Phenotypic expression of VHL disease is also influenced by genetic modifiers.26
Intragenic mutations and partial deletions result in a typical disease presentation, while complete gene deletions have not been extensively studied and an association between complete VHL gene loss and disease phenotype has not been exhaustively investigated. Recently, in one Belgian family with a deletion of the entire VHL gene, only cHAB without rHAB and RCC were diagnosed.27 For genetic counselling purposes, it is important to determine whether patients with complete deletions have reduced disease expression.
So far, germline VHL mutations have not been investigated in any population of Slovenian origin. We report the results of a study of 34 Polish families analysed in order to describe more precisely: (1) the frequency of constitutional mutations in patients matching the clinical criteria for VHL; (2) the spectrum of germline VHL mutations in the Polish population; (3) the proportion of germline de novo mutations; and (4) genotype-phenotype correlations in patients with deletions of the entire VHL gene.
MATERIAL AND METHODS
Families
Thirty-four unrelated families diagnosed on the basis of clinical criteria were tested for germline mutations in the VHL gene. The families came from medical centres in different regions of Poland. Complete clinical information, including ophthalmological, intracranial, and visceral findings were collected from 82 patients with the disease phenotype. Thirty-two families presented without phaeochromocytoma (VHL type 1) and two families with phaeochromocytoma (VHL type 2).
DNA isolation
DNA was isolated from peripheral blood leucocytes using proteinase K digestion and phenol/chloroform purification. Aliquots of DNA were dissolved in sterile distilled water to a working concentration of 50 ng/μl.
Sequencing
The entire coding sequence of the VHL gene was amplified in a PCR reaction using primer pairs 3, 101, 102, 103, and 6, 107.28 After purification, PCR products were sequenced with the same primers, using fluorescently labelled dideoxy chain terminators from an ABI Prism kit (Applied Biosystems) in an ABI 373 automated sequencer.
Long PCR
Screening for large deletions was initially performed using long range PCR reaction with three overlapping pairs of primers as described previously.29
Multiplex PCR
Cases negative by long range PCR were investigated by universal primer quantitative fluorescent multiplex polymerase chain reaction (UPQFM-PCR). This method is an adaptation of the UPQFM-PCR technique developed from detection of rearrangements in the LDL-R gene.30 Three exons of the VHL gene and a fragment of the β-globin gene (internal control) were amplified in two step PCR. Detailed experimental conditions and applied primers are shown in table 1. Two μl of final product were mixed with 2 μl of dextran blue formamide loading dye, denatured at 94°C for four minutes and 2 μl of each sample was electrophoresed in a 36 cm 6% denaturing acrylamide gel. Detection of large deletions was based on comparison of peak areas of exons of the VHL gene and of a β-globin fragment amplified in multiplex PCR. Peak areas were calculated using GeneScan Analysis software.
Oligonucleotides (A) and conditions (B) for UPQFM-PCR
Quantitative Southern analysis (performed by Italian coworkers)
For Southern analysis, DNA was digested with EcoRI and AseI and hybridised with the VHL g7-cDNA probe and a β globin probe as described previously.9
RESULTS
Germline mutations
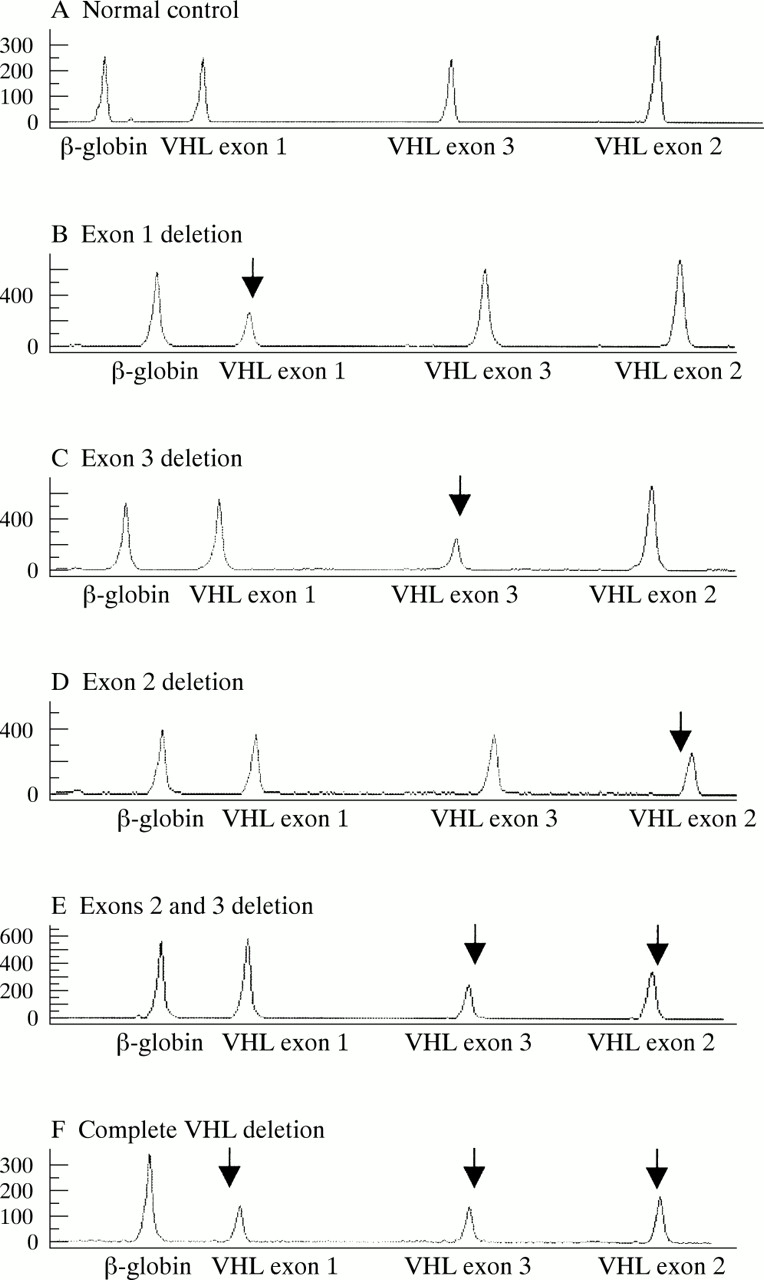
Sequencing detected mutations in 18 families. In five families, partial gene deletions were detected by long range PCR, as reported previously.29 In seven of 11 probands negative by sequencing and long range PCR, two partial deletions and five complete VHL deletions were detected by means of multiplex PCR (the peak patterns of the UPQFM-PCR products characteristic for five different deletions are shown in fig 1). Six of 11 patients investigated by multiplex PCR were also tested by QSA and the same results were obtained (partial deletions in two patients, complete deletion in two patients, and a failure to detect mutation in two cases (table 2)).
Germline mutations in the VHL gene and related phenotypes in 82 patients from 34 unrelated Polish families fulfilling clinical VHL criteria
{kind=link}
Genotyper traces of a normal control and five deletions. Peaks corresponding to deleted exons are marked by arrows. Peaks are labelled by the name of the amplified region.
Altogether, germline VHL mutations were detected in 30/34 (88%) probands (table 2). We observed 12 different intragenic mutations in 18/30 (60%) families: six missense mutations (10 families) and one nonsense mutation (one family), one in frame deletion (three families), four frameshift mutations, three deletions (three families) and one insertion (one family). All intragenic mutations were localised between nucleotides 194 and 500 of the coding sequence: seven different mutations (12 families) were found in the second half of exon 1, one mutation (one family) in exon 2, and four mutations (five families) in the 5` part of exon 3. Recurrent VHL changes were found in 13/18 (72%) families with VHL mutations detected by sequencing. In 10 of them, four different mutations were identified (194C>T, 226_228delTTC, each in three families, and 233A>G, 500G>A, each in two families). Three mutations (333C>G, 481C>T, 485G>A) were not recurrent in our patients, but the same changes were reported in VHL patients from other populations. We observed five mutations (233A>T, 217delC, 263_265delGGCinsTT, 445delG,477_478insCA) unique to our population. Large deletions were detected in 12/30 (40%) families, including 7/30 (23%) partial deletions and 5/30 (17%) deletions of the entire VHL gene. Partial deletions were heterogeneous in size and position: one removed exon 1, two deletions were restricted to the central exon, four deletions involved the 3` end of the gene. Haplotype analysis using polymorphic DNA markers D3S1038, D3S1317, and D3S65131,32 was performed to determine whether the high proportion of subjects with a complete deletion was the result of a founder effect. A different haplotype was found in families 26, 28, 29, families 27 and 30 showed a common haplotype, but a proband from family 30 showed an apparent de novo mutation, so there was no evidence for founder effect for complete deletions in our population (table 3).
Clinical presentation and haplotype analysis in patients with a deletion of the entire VHL gene
Five of 30 (17%) mutation probands were found to have no family history of VHL disease. In two cases, samples from both parents were available and germline mutations (233A>G substitution, ∼10 kb deletion of exons 2 and 3) were not detected in DNA extracted from blood leucocytes of the parents.
Mutations were not detected in four probands (five persons/four families) fulfilling clinical criteria for VHL: a male diagnosed with cerebellar HAB at the age of 16 and multiple spinal HABs at the age of 19 and his brother with cerebellar HAB at the age of 19; a girl with cerebellar and spinal HABs at the age of 9 years; a man with multiple spinal HABs at the age of 14; and a female with rHAB and cerebellar HAB at the ages of 13 and 17, respectively. In four of the patients with a negative result of VHL gene analysis, diagnosis of cHABs was confirmed by histopathology of tumour tissue. In the female with rHAB, the cerebellar haemangioblastoma was diagnosed by MRI. In order to identify a possible somatic mosaicism, the mutation negative patients were additionally tested by SSCP analysis (performed with primers described previously28). The analysis did not show somatic mosaicism in the patients.
Phenotypes in patients with complete gene deletions (table 4)
Clinical presentation in patients with complete deletions and other VHL type-1 mutations
Of 11 patients with null mutations, all presented with cHAB diagnosed at a mean age of 32 (range 20-62). Cerebellar and spinal HABs were observed in 11/11 (100%) and in 4/11 (36%) patients, respectively. cHABs were symptomatic at the time of diagnosis in 10/11 subjects. Histopathology of tumour tissue confirmed the diagnosis of HABs in these cases. In 1/11 patients, asymptomatic cHAB was identified by MRI at the age of 20. Two out of 11 (18%) subjects developed retinal angiomas. One of them developed a single symptomatic angioma of the optic disc at the age of 30. The second was affected by asymptomatic bilateral retinal angiomas diagnosed at the age of 30. Multiple renal cysts were observed in 2/11 (18%) patients. A single renal cyst was diagnosed in 1/11 (9%) subjects with complete deletions.
In order to find possible distinct features of VHL disease in patients with complete deletions representing true null alleles, we compared the phenotype in these patients with the phenotype in subjects with other VHL type 1 mutations (VHL type 1 missense mutations, protein truncating mutations, partial gene deletions). Patients with complete deletions differed from the ones with other VHL type 1 mutations. They rarely developed rHAB compared to patients with intragenic mutations and partial deletions (2/11 (18%) versus 45/57 (80%)); they commonly developed cHAB (11/11 (100%) versus 47/57 (82%)), and were not affected by RCC in contrast to those with other mutations (16/57 (28%)).
DISCUSSION
In the present study, germline VHL mutations were detected in 30/34 (88%) unrelated Polish patients with a clinical diagnosis of VHL. Although we used recently developed methods of analysis of the VHL gene, we failed to identify germline mutations in four (12%) probands, who exhibited predominantly early onset brain tumours and/or retinal angioma. Recently also, Hes et al,33 using similar analyses, did not detect germline VHL mutations in two (out of four) patients with multiple cHAB at the ages of 44 and 66.
It is of interest whether the mutation negative patients with a clinical diagnosis of VHL have VHL disease. If they are affected by VHL, they may represent cases of somatic mosaicism, the condition which is defined as the presence in a person of at least two cell lines differing in genotype and arising from a single zygote.34 Mosaicism could account not only for mutation negative patients with a clinical diagnosis of VHL, but also for the mutation negative parents of a patient with an apparent de novo mutation. Such a phenomenon may be relatively common, since it has been shown that at least two (4.8%) of 42 parents of subjects with apparent de novo mutations were somatic for VHL.21 Patients somatic for a mutation tend to have less severe disease than their non-mosaic relatives, but this may not always be the case.34 Of three patients with VHL mosaicism confirmed by DNA analysis, two had subclinical disease and one presented with advanced, symptomatic VHL.21,35 In this context, not only patients with late onset disease, but also those with early onset may be somatic for VHL.
A mutation is present only in some cells or tissues of a somatic subject and it may not be detected by standard testing methods such as sequencing and Southern blotting. Thus, we checked for mosaicism using SSCP analysis on DNA extracted from blood leucocytes, as this material was available for study. SSCP could detect mosaicism for a single base change. However, FISH would be necessary to detect mosaicism for a complete deletion. Also, analysis of DNA from other cells or tissues (such as cheek epithelium or skin fibroblasts) might prove useful for detecting a mutation present only in some cells or tissues of the body. In addition, the offspring of a mutation negative patient with a clinical diagnosis of VHL may be tested for a germline mutation, as the affected children of a mosaic subject have regular disease.
If the mutation negative patients have VHL, their mutations may be located elsewhere or be of a different nature from the mutations for which the assays are designed. It is interesting that these patients presented primarily with brain tumours, the phenotype that would suggest a complete deletion of one VHL allele (or a null allele). A null allele might also result from silencing of the VHL gene caused by deletions or point mutations in 5` or 3` of the gene that affect gene expression or mRNA stability, mutations in the introns that affect mRNA stability or splicing, or abnormal methylation. A quantitative RT-PCR analysis of the VHL mRNA in lymphocytes might indirectly identify such mutations, and could help to answer the question whether the mutation negative patients truly have VHL.
If the mutation negative patients are not affected by VHL, they may be phenocopies (independent mutation events giving rise to various HAB could also be expected by chance), especially those with later age at onset. The failure to detect germline VHL mutations in patients with HAB only may also suggest the presence of additional haemangioblastoma susceptibility genes. For example, defects in elongin B, C, Cul 2, or Rbx1 (which form a multimeric complex with pVHL (the VCBCR complex)), or HIF transcription factors (which are regulated by this complex) could be involved in the pathogenesis of HAB. However, recent studies suggest that pVHL may be the sole mutational target through which the VCBCR complex is disrupted in RCC,36 but this question was not investigated in HAB. Also, mutations in other genes regulated by the VCBCR complex might be associated with HAB development.
This is the first description of germline VHL mutations in patients of Slovenian origin. The characteristics of mutations in Polish families are similar to those reported in families from other populations with regard to localisation, types of germline VHL mutations, and occurrence of common mutations. We observed 18 intragenic mutations, 10 missense and eight non-missense mutations (nonsense, in frame and frameshift microdeletions, or microinsertions). The same types of intragenic mutations were detected in VHL patients from other populations. All the intragenic mutations were localised between codons 65-167; all of them mapped to the highly conserved sequence downstream from the second methionine codon (codon 53). This conserved region encodes two functional VHL domains, the α and β domains. The VHL mutations map to both domains and it was suggested that two intact macromolecular binding sites may be required for the effects of VHL protein.22 So far, the non-conserved coding VHL region localised upstream of the second methionine was not found to be altered by any disease causing mutations. In the non-conserved coding region, we detected only the 74 C>T (proline 25 to leucine) polymorphism in three unaffected members of Polish VHL families. This 74 C>T polymorphism was also detected in families from North America and Europe.13,37
The collection of a large number of mutations is necessary to identify common DNA changes and is important in showing key residues in the protein function. Data on a total of 137 different mutations in VHL families from North America, Europe, and Japan, collected by Zbar et al,15 enabled identification of six common germline VHL mutations, which were observed in four or more VHL families, in contrast to most germline mutations, found in one or two families.15 We identified the same common VHL mutations in Polish families (233A>G, 226-228delTTC, 481C>T, 500G>A). A C>T substitution at nucleotide 194, detected previously in three families from UK, France, and Germany, was also detected in three unrelated probands of the present study, making this a relatively common germline mutation. Like most common VHL mutations it is localised in hypermutable CpG dinucleotides.
Of the 30 unrelated probands with germline VHL mutations, we identified partial VHL deletions in seven (23%) patients. This frequency is similar to those reported by studies from North America and England. A lower prevalence was observed in Japanese (5%). Partial gene deletions detected in our series were 1-10 kb in length, removed exon 2 or the 5` or 3` ends of the gene, and were similar to those identified by Richards et al,38 who characterised a large series of 22 partial deletions of the VHL gene.38
Previously, deletions of the entire VHL gene investigated by PFGE were found with a low frequency of 3%.39,40 The frequency of deletions of the entire VHL gene detected by quantitative Southern analysis in the study of families from the USA was 8/92 (9%), and in a separate group of VHL patients, deletions of the entire VHL gene were identified in 3/50 (6%) probands.9 Complete gene deletions were observed in as many as five (17%) probands from Poland. According to haplotype analysis such a high proportion is not the result of founder effect in our population. Varying prevalences of large deletions may reflect ethnic differences in localisation and nature of the sequences (that is, homologous sequences or Alu repeats) known to promote deletions to occur.
Similar mutation associated phenotypes were reported in VHL families of different origin (with a minor exception of a few mutations associated with VHL type 1 in western VHL, which were associated with VHL type 2 in Japanese families).15,18,41 In agreement with this, in VHL type 1 families, we identified protein truncation mutations, large deletions, and specific missense mutations, also not associated with development of phaeochromocytoma in families from other populations. Non-missense mutations predominated (20/28, 71%) in Polish type 1 families, as in type 1 families from North America, Japan, and western Europe. Specific missense mutations were associated with a risk of phaeochromocytoma, particularly two missense mutations, arg167trp and arg167gln, conferred a high (62%) risk for phaeochromocytoma. In line with this observation, phaeochromocytoma was diagnosed in all our families with arg167gln mutation. The positive correlation between specific missense mutations and phaeochromocytoma is useful in genetic counselling. VHL kindreds with such missense mutations require careful surveillance for phaeochromocytoma as this is frequently the first manifestation.14
Recently, in five members of a Belgian family with complete VHL gene deletion, cHABs without rHAB, RCC, and phaeochromocytoma were diagnosed.27 Apart from this report, phenotypes in patients with complete deletions have not been published. In the present study, we characterised the clinical presentation in five unrelated families with loss of the entire VHL gene. Of 11 patients with null mutations, all presented with cHAB diagnosed at a mean age of 32 (which is the mean age of HAB diagnosis in VHL disease), suggesting that complete deletions are associated with a high risk of cHAB. In contrast to members of the Belgian family, two out of 11 (18%) of our patients developed retinal angiomas, suggesting that complete deletions also confer a risk of rHAB. However, this frequency is lower than the prevalence of retinal lesions in VHL patients with intragenic mutations and partial deletions, which is 80% in our series and about 45-59% in other series.41 In addition, rHAB was the first disease manifestation in only one of 11 patients with complete deletions, while rHAB is usually diagnosed earlier than cHAB in VHL patients. Apparently, complete deletions are associated with a relatively low risk of retinal angiomatosis. Renal cell cancer has not been diagnosed in the Polish and the Belgian patients with complete deletions, at a mean age of 38 and 57 years, respectively. The age related risks of renal cell carcinoma in subjects with intragenic mutations and partial gene deletions are about 20% and 60% at the same ages, respectively.14 Thus, complete deletions may be associated with a low risk of RCC.
It is interesting why most patients with a complete deletion of one VHL allele exhibited a HAB only phenotype. The pathogenesis of tumours involves inactivation of one VHL allele by a germline mutation and a second “hit”, which is frequently loss of heterozygosity (LOH), but may be abnormal methylation or a small intragenic mutation.42 In cells with a constitutional complete deletion of VHL, LOH is predicted to cause true null VHL alleles. It is possible that the true null alleles are not sufficient for development of all VHL related lesions, are primarily associated with development of cHAB, and as LOH occurs frequently, patients with complete deletions exhibit predominantly brain tumours. In this model, a typical VHL disease presentation in patients with a constitutional null allele could be associated with less common types of somatic inactivation, such as by small intragenic mutation (this mechanism might have a similar effect to LOH in a patient with a germline intragenic mutation, known to result in the VHL type 1 phenotype). Analysis of a type of somatic inactivation in tumours of patients with complete deletions might help to verify this hypothesis. However, the pathogenesis of tumours also involves inactivation and/or activation of other genes (for example, VHL inactivation is not sufficient for RCC tumorigenesis, and inactivation of 3p12-p21 tumour suppressor genes (such as RASSF1A) appears to be necessary in RCC irrespective of VHL gene inactivation status43). In this context, the complete deletion related phenotype could also be attributed to the fact that complete deletions might remove some important sequences (or genes) near the VHL gene. Also, the presence of intact VHL protein (in cells with the constitutional loss of one VHL allele, only the wild type one is expressed) could provide a measure of protection against disease and be in a part responsible for the observed phenotype.
Key points
-
Thirty-four Polish families with a clinical diagnosis of VHL disease were studied in order to describe: (1) the frequency of germline mutations in these families; (2) the spectrum of germline VHL mutations in the Polish population; (3) the proportion of de novo mutations; and (4) genotype-phenotype correlations in patients with a deletion of the entire VHL gene.
-
The coding region of the VHL gene was tested using direct sequencing. Large deletions were analysed using quantitative Southern blotting and/or multiplex PCR.
-
(1) Germline VHL mutations were observed in 30/34 (88%) families. Mutations were not identified in 4/34 (12%) probands (five subjects/four families) with central nervous system haemangioblastoma (cHAB) and/or retinal angioma (rHAB). (2) Small intragenic mutations were detected in 18/30 (60%) families; all were located 3` of codon 53. Partial and complete gene deletions were detected in 7/30 (23%) and 5/30 (17%) families, respectively. Five mutations were unique to the Polish population. (3) Five of 30 (17%) VHL mutation positive probands were found to have no family history of VHL. (4) Of 11 patients with complete deletions, all developed cHAB, two presented with rHAB, and none developed renal cell carcinoma (RCC).
-
(1) Some patients with predominantly brain tumours and/or retinal angioma do not have identifiable germline mutations in the VHL gene and may have somatic mosaicism, or may be affected by mutations of different nature or localisation than the mutations for which the study assays are designed, or may be phenocopies of the disease. An alternative explanation is the presence of additional haemangioblastoma susceptibility genes. (2) The main characteristics of germline VHL mutations in the Polish population are similar to those reported in other populations. However, we observed a higher proportion of patients with complete deletions (5/30, 17%), compared to those reported in other populations (3-9%). There was no evidence of a founder effect for complete deletions in our patients. (3) The apparent de novo mutation rate is ∼20%. (4) A complete deletion of the VHL gene results primarily in brain tumours. This result may be useful in genetic counselling for subjects with complete deletions.
VHL kindreds with complete deletions require careful surveillance for cHAB, as this is common and most frequently the first manifestation. Although deletions of the entire VHL gene may be associated with a relatively low risk of rHAB, surveillance for retinal lesions should be performed in patients with complete deletions, as some of them will develop rHAB. RCC was not observed in any of 16 subjects with complete deletions, but it cannot be ruled out that complete deletions confer a risk of RCC; therefore, VHL patients with such mutations should continue to be monitored for the presence of RCC. It is possible that complete deletions confer a low risk of RCC. As renal cell cancer is a common cause of death in subjects with VHL, complete deletions may be associated with a better prognosis for life expectancy than other types of VHL mutations. Because of the clinical significance of possible reduced expression of the disease in patients with deletions of the entire VHL gene, it is necessary to verify our observations. Particularly, it will be very important to investigate a risk of RCC in subjects with complete deletions in a larger series of cases.
Acknowledgments
This work was supported by grant KBN No 4P05A 130 18 from the State Committee for Scientific Research, Warsaw, Poland. We thank the following for cooperation in founding the Polish VHL Registry: M Zalewska, A Stankiewicz, R Sujkowska, A Domaniewski, G Malukiewicz, J Kaluzny, B Wysocka, H Dembicka, K Raczynska, E Starzycka-Bigaj, K Chrzanowska-Srzednicka, D Chibowski, M Czochara, W Papierz, W Paprzycki, M Slojewski, R Kaczor, E Wlodarczyk, D Karczewicz, M Modrzejewska, A Palacz, W Lubinski, W Poncyliusz, K Krzyslak, Z Domanski, G Wilk, B Górecka, G Psut, J Kubalska, A Kukwa, S Józwiak, A Borkowski, H Pytrus-Zajac, H Nizankowska, and A Lubinska. We thank Professor R Scott for critical improvements.