Article Text

Statistics from Altmetric.com
Rett syndrome1 is a neurodevelopmental disorder that primarily affects females and is characterised by a period of normal growth and development, followed by severe neurological dysfunction including dementia, autistic features, loss of purposeful use of the hands, jerky truncal ataxia, and seizures.2 Systematic genetic analyses have shown that mutations in the methyl-CpG binding protein gene MeCP23 are associated with Rett syndrome.4 Recently, MeCP2 mutations (A140V5,6 and E137G6) were also found in male patients with non-specific X linked mental retardation that is clinically distinct from Rett syndrome. To elucidate the functional significance of these mutations, we used transient expression assays to compare the effects of these mutations on MeCP2 function with those of Rett syndrome mutations.4 Wild type and mutant MeCP2/GFP fusion proteins expressed in mouse L929 cells were analysed to determine the effect of mutations on accumulation of MeCP2 in heterochromatin, where approximately half of the methyl-CpG dinucleotides occurring in the genome are located.7 In contrast to an R106W Rett syndrome mutant protein, which has no affinity to heterochromatin, both A140V and E137G mutants showed a clear focal heterochromatin staining pattern indistinguishable from the wild type protein. The effects of mutations on transcriptional repressive activities were also evaluated in Drosophila SL2 cells, which possess only marginal background activities of methyl-CpG binding proteins.8–10 Although R106W expression substantially reduced transcriptional repressive activity, the A140V and the E137G mutants retained the transcriptional repressive activity. In particular, the A140V mutant retained such repressive activity at a level comparable to the wild type protein. These results indicate that potential alterations in MeCP2 function resulting from the A140V and the E137G mutations are different from those associated with mutations observed in Rett syndrome and may explain why the manifestation of MeCP2 related mental disorder in males is clinically different from Rett syndrome.
MATERIALS AND METHODS
MeCP2 cDNA bearing missense mutations were generated by site directed mutagenesis using mismatched primers as described previously.11 A full length MeCP2 cDNA12 was used as a template for PCR. Insert of the A140V5,6 or E137G6 mutant DNA was cloned into the BspEI and XhoI sites of pEGFP-C1(Clontech), an enhanced fluorescence vector, and the EcoRI and BamHI sites of the Drosophila expression vector pAc5.1/V5-His (Invitrogen). Mouse L929 cells were transfected with GFP expression constructs using Superfect (Qiagen). Two days later, cells on chamber slides were fixed with 3.7% formaldehyde for 10 minutes, permeabilised with 0.5% Triton X-100 for 20 minutes, and counterstained with 4`,6-diamidino-2-phenylindole (DAPI) (Sigma). The specimens were observed under an Olympus fluorescence microscope using the appropriate optical filter. The transient expression analysis using Drosophila SL2 cells was performed as follows. The SNRPN-luciferase reporter construct13 was treated with SssI (CpG) methylase (New England Biolabs) in the presence of 5 mmol/l S-adenosylmethionine as described previously.14 A total of 1.4 × 105 SL2 cells were grown in 0.7 ml of Schneider's Drosophila medium in a 24 well plate. A total of 0.4 μg of the luciferase reporter construct was cotransfected with 0.2 μg of an Sp1 expression plasmid, pPacSp1, 0. 01 μg of pAc5.1-pRL,15 and various amounts of expression plasmid (1-100 ng) bearing genes encoding MeCP2 mutants into Drosophila SL2 cells by the calcium phosphate method. The total amount of transfected DNA was adjusted by adding pAc5.1/V5-His vector. After 48 hours, the cells were lysed with 100 μl of lysis buffer and 10 μl of lysate was assayed for firefly and Renilla luciferase activities using the Dual-Luciferase reporter assay system (Promega). All transient transfection assays were carried out at least three times independently.
RESULTS
Recently, novel mutations in the methyl-CpG binding domain (MBD) of MeCP2 were reported in males affected by a mental retardation disorder that is clinically distinct from Rett syndrome.5,6 The E137G mutation was found in a family in which affected males showed profound to mild mental retardation, often associated with speech handicap.6 The A140V mutation was found in a family, four adult, severely mentally retarded brothers, with mild mental retardation in their mother and sister.5 The A140V mutation was also found in sporadic cases of moderate to severe mental retardation in males.6 It has been assumed that males who are hemizygous for mutations in MeCP2 die prenatally or early in infancy with a severe congenital encephalopathy. Therefore, survival of male patients exhibiting mental retardation into adulthood suggests that the activities of these novel mutant proteins are functionally distinct from that of MeCP2 mutant proteins seen in Rett syndrome. In order to investigate the functional consequences of the A140V and E137G mutations, we used a heterochromatin staining analysis, which was initially used to characterise Rett syndrome mutations.16 This analysis takes advantage of the unique feature of mouse L929 cells, which have approximately half of the methyl-CpG dinucleotides clustered in the pericentromeric heterochromatin.7Thus, exogenously expressed MeCP2 accumulates in this region and can be detected by fluorescence microscopy as distinct foci.3,17 Such accumulation of MeCP2 in heterochromatin is not observed in human or rat cells.3 Loss of MBD function leads to a decrease in the intensity of focal staining and an increase in staining throughout the nucleus.16 Therefore, the effect of MBD missense mutations on methyl-CpG binding activities can be indirectly assessed. Using this system we compared the activities of the A140V and the E137G mutant proteins with that of wild type protein and with two common Rett syndrome mutations, R106W and T158M.4 A previous study showed that fusing MeCP2 mutant proteins with GFP did not affect their nuclear localisation,16 so mutants were expressed as GFP fusion proteins. Mouse L929 cells were transfected with GFP fusions of wild type and mutant proteins, and intranuclear localisations of proteins were examined. The result of this analysis is summarised in fig 1. MeCP2-GFP was predominantly localised to heterochromatin regions of mouse L929 cells, which were identified by co-localisation of strong DAPI staining. As reported previously,16 R106W-GFP showed no clear focal staining in the nucleus and fusion proteins were distributed throughout the nucleus. Impairment of heterochromatin staining was also observed with the T158M-GFP construct, although the effect was milder than that of R106W. Some focal staining in a background of nuclear staining is still apparent. By contrast, both A140V-GFP and E137G-GFP fusion proteins showed the distinct foci in the nucleus, which matched with DAPI staining, indicating that the mutant proteins were predominantly localised to heterochromatin. These staining patterns are indistinguishable from that of the wild type protein, indicating that the A140V and the E137G mutants retain substantial affinities for heterochromatin and their abilities to bind methyl-CpG are not obviously affected.

Heterochromatin staining with GFP-MeCP2 mutant proteins. Mouse L929 cells were transiently transfected with constructs expressing GFP fusion protein with MeCP2 (wt) or mutants bearing missense mutations as indicated. After 48 hours, the cells were fixed and permeabilised, and the nucleus was counterstained with DAPI. Intranuclear localisation of GFP fusion proteins was visualised as described in Materials and methods. The same staining pattern was observed in almost all fluorescence stained cells in any field.
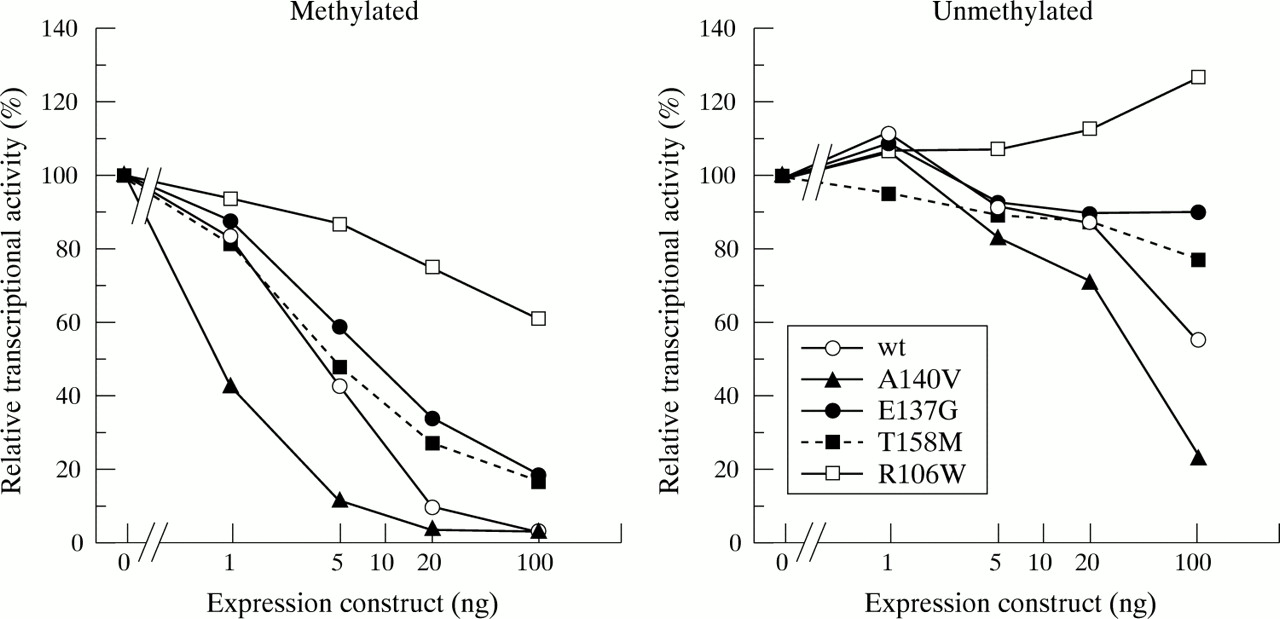
To delineate the effects of the A140V and the E137G mutations on the transcriptional repressive activity of MeCP2, we used transient expression in a Drosophila cell line. It is known that Drosophila cells have very low levels of DNA methylation18,19 and exhibit extremely low levels of the DNA methyltransferase activity8,20 and methyl-CpG binding activities.8–10 These observations suggest that regulation by DNA methylation is negligible in Drosophila cells. On the other hand, the general transcriptional machinery is thought to be highly conserved between Drosophila and mammals.21 Our previous studies indicated that wild type MeCP2 expressed in Drosophila SL2 cells repressed Sp1 activated transcription on a methylated promoter,14 indicating that SL2 cells are suitable to evaluate MeCP2 repressive activity. Here, we used a construct containing the luciferase reporter driven by the promoter of the human imprinted SNRPN gene. This SNRPN promoter provides high transcriptional activity and is highly sensitive to MeCP2 transcriptional repression.13,15Drosophila expression constructs encoding wild type or a mutant MeCP2 were cotransfected into SL2 cells with the reporter plasmid and an Sp1 expression vector, as well as the pAc5.1-pRL control plasmid for normalisation. The results of this analysis are shown in fig 2. As shown previously,16 wild type MeCP2 sharply reduced Sp1 activated transcription in a dose dependent manner. Transcriptional activity was reduced approximately 30-fold at the highest dose (100 ng). On the other hand, cotransfection with R106W had little effect on transcription from the methylated template; transcription was repressed approximately 40% at the highest dose, indicating that R106W mutant protein significantly lacks repressive activity.
{kind=link}
{kind=link}
The unmethylated or methylated SNRPN-luciferase construct (0.4 μg) was cotransfected with pPacSp1 (0.2 μg), a Renilla luciferase vector pAc5.1-pRL (0.01 μg), and various amounts of expression plasmids (1-100 ng) encoding wild type or mutant MeCP2 into Drosophila SL2 cells. The total amount of transfected DNA was adjusted by adding pAc5.1/V5-His vector. Relative transcriptional activities compared with that of the pAc5.1/V5-His vector only are presented. Renilla luciferase activity was used for normalisation.
Intermediate levels of transcriptional repression were seen with the T158M mutant. Transcriptional repressive activity of T158M was slightly stronger than that of R106W, showing five-fold repression of transcription at the highest dose. The E137G mutant showed the transcriptional repressive activity similar to that of the T158M mutant when the promoter was methylated. On the other hand, the A140V mutant strongly repressed transcriptional activity, showing a steep dose response curve compared to that of the wild type protein. Thus, the A140V mutant retains substantial repressive activity on a methylated promoter. Interestingly, the ability of the A140V mutation to repress transcription from an unmethylated SNRPN promoter was also significantly enhanced, reducing luciferase activities by approximately five-fold at the highest dose (100 ng), while the wild type protein reduced transcription by only about 40% at the same dose. In this study, we also examined expression levels of expressed wild type and mutant MeCP2 proteins by indirect immunofluorescence and western blot analysis, and found that the proteins expressed from these plasmids were produced in the nucleus at similar levels following transfection (data not shown). Table 1 summarises these two transient expression analyses. Based on these results, the A140V and the E137G mutants retain more residual function in terms of heterochromatin binding and transcriptional repression than two common Rett syndrome mutants. This observation may explain why male patients carrying these novel mutations survive into adulthood with mental retardation, a phenotypic manifestation different from that seen in Rett syndrome.
Summary of the effects of mutations
DISCUSSION
MeCP2 mutations were initially found in patients with Rett syndrome, which occurs almost exclusively in females.4 Mosaic expression of normal versus abnormal alleles resulting from X chromosome inactivation probably contributes to the survival of female patients and also the severity of Rett syndrome,22 while males who are hemizygous for mutations in MeCP2 most often present with a severe congenital encephalopathy associated with decreased survival.23 Similarly, MeCP2 null mice and mice undergoing conditional knockout of MeCP2 before birth can survive and develop symptoms characteristic of Rett syndrome after weaning.24,25 The recent findings of MBD mutations in adult males with non-specific X linked mental retardation5,6 suggest that the altered activities of these mutant MeCP2 proteins are responsible for these phenotypes. To address this question, the functions of these novel mutant proteins were examined by two assays and the results were compared to similar assays with known Rett syndrome mutant proteins. A previous study showed that most missense mutations in MBD of MeCP2 substantially reduced the affinity of MeCP2 for heterochromatin in L929 cells.16 These mutations also abolished the repressive activity of MeCP2 on Sp1 activated transcription from a methylated promoter.16 Mouse heterochromatin is known to contain satellite DNA, which possesses highly methylated CpG dinucleotides.7 It has been shown that the accumulation of MeCP2 in mouse heterochromatin requires both methylated CpG and the MBD.17 Therefore, the accumulation of MeCP2 in heterochromatin depends primarily on the ability of MBD to bind to methylated DNA. Heterochromatin staining analysis of L929 cells showed that A140V-GFP and E137G-GFP fusion proteins exhibited clear focal staining in the nucleus, a pattern similar to that seen in the wild type protein, indicating that the binding affinities to methylated CpGs are not decreased by these mutations. Transient transfection of Drosophila cells also enabled us to evaluate the consequences of these novel mutations on MBD function. A previous study showed that C-terminal truncated MeCP2, which contains the MBD but not the transcriptional repression domain (TRD), exhibited substantial transcriptional repressive activity in Drosophila cells.12 Therefore, it is likely that transcription is repressed primarily by steric hindrance induced by MeCP2 binding to methylated CpGs, making the promoter inaccessible to activators. However, in this system, active transcriptional repression26,27 may also be partially functional owing to interaction between the TRD and histone deacetylase complex. In fact, the histone deacetylase inhibitor tricostatin A (TSA) relieved the transcriptional potential and the efficiency of recovery varied depending on the promoter context (data not shown). Since binding of the MBD to methylated DNA is a prerequisite for recruitment of histone deacetylase, the effect of the MBD mutation on transcriptional repression can be assessed in our Drosophila system. Transient expression of the A140V mutant in Drosophila SL2 cells showed that this mutant retained considerable ability to repress Sp1 activated transcription from a methylated reporter. Intriguingly, the A140V mutant showed stronger repression of transcription from an unmethylated reporter than did the wild type protein. It is noteworthy that the R106W mutant, which had low affinity to heterochromatin, completely failed to repress Sp1 activated transcription from an unmethylated template (fig 2). It has been shown that the region flanking the carboxy side of MBD is involved in binding to unmethylated DNA.28 Our transcription analysis in SL2 cells suggest that in addition to that region, the MBD itself contains a region involved in non-specific binding to unmethylated DNA. Although further studies are required to show the significance of MeCP2 affinity to unmethylated DNA, it is possible that higher affinity binding owing to the A140V mutation affects the proper gene expression although methylated genes are normally controlled. NMR spectroscopy analyses have shown that an alanine at amino acid residue 140 is present in the middle of the α helix of MBD.29,30 Therefore, a mutation in this locus may not significantly change the folding of the MBD structure and would barely perturb its binding to methyl-CpG base pairs; however, such a mutation may increase the affinity for unmethylated regions. Although we cannot rule out the possibility that the A140V mutation is not a disease causing mutation but just a polymorphism, transcriptional repressive effect of this mutation on the unmethylated gene is apparently different from that of wild type. This change of the MeCP2 function may result in an aberrant pattern of gene expressions in relevant tissues. On the other hand, the E137G mutation exhibited further impairment of the transcriptional repressive function in our Drosophila assay, even though this mutant possessed the mouse heterochromatin affinity comparable to that of wild type. The effect of the E137G mutation on the transcriptional repressive activity to the methylated promoter was almost equal to that of the T158M mutation (fig 2). Recently, the T158M mutation was observed in two brothers of a patient with classical Rett syndrome.31 Both brothers died within a year of birth, and one suffered from severe encephalopathy.31 Our functional analyses show that T158M had intermediate affinity to heterochromatin and moderate effects on transcriptional repressive activity. These results are consistent with reports that the T158M mutant exhibited only slightly lower binding affinity for methylated DNA compared with that of the wild type protein.32,33 Thus, mild impairment of MeCP2 function owing to mutations may result in MeCP2 associated mental retardation or severe encephalopathy in males. Taken together, these observations suggest that the MeCP2 mutations give rise to a variable range of neurodevelopmental disorders dependent on the functionality of the protein.
-
Mutations in the MBD of MeCP2 (A140V and E137G) were recently found in male patients affected with non-specific X linked mental retardation.
-
We have characterised these mutations by comparing them with the common MBD mutations found in Rett syndrome patients.
-
The functional significance of the mutations was analysed by transfection in mouse L929 cells and Drosophila SL2 cells to investigate the effects of mutations on the affinity to heterochromatin and the transcriptional repressive activity, respectively.
-
The A140V and E137G mutations exhibited mild impairment of MeCP2 function, which may be implicated in the manifestation of MeCP2 related mental disorder in males.
Acknowledgments
We thank Drs Kei Fujinaga and Takeo Kubota for helpful discussions and Dr Ellen E Lamar for critical reading of the manuscript. This work was supported by the Akiyama Foundation.