Article Text

Statistics from Altmetric.com
Editor—Using linkage analysis, a large Indian family with autosomal dominant sutural cataract and cerulean opacities was mapped to chromosome 22 and two cosegregating sequence changes (475C→T and 483C→T) were identified in theCRYBB2 gene. The first was previously described in two genetically unrelated families with other inherited forms of cataract. The two sequence alterations are identical to the sequence of the CRYBP1 pseudogene that is 228 kb apart. Furthermore, the pseudogene-like fragment within theCRYBB2 gene is flanked by chromosomal junction sequences. Therefore, we conclude that gene conversion is the most likely mechanism leading to this mutation. Alternatively, dual point mutation would explain our findings. In addition, since the three families with Q155X mutations all show different types of cataract, we conclude that mutant CRYBB2 causes cataract formation but other modifying factors determine the type of cataract.
Autosomal dominant congenital cataract (ADCC) is a clinically and genetically heterogeneous group of disorders that cause blindness. More than 13 independent loci have been mapped, and 10 different genes identified so far. Five of them are crystallin genes that are categorised into the α, β, γ, μ, and ζ subgroups. The crystallins constitute the main lens proteins, whereby β-crystallin B2 is the only abundant protein in the adult lens fibre in man.1 ,2 Causative mutations have been recognised in the α-crystallin A gene (zonular central nuclear cataract),3 the β-crystallin A3/A1 gene (zonular cataracts with sutural opacities),4 the γ-crystallin C gene (Coppock-like cataract),5 and the γ-crystallin D gene (progressive juvenile onset punctate cataract).6These and all other ADCC mutations identified so far are private mutations, with one exception. Litt et al 7 described a nonsense mutation, Q155X, in the β-crystallin B2 gene leading to cerulean cataract. Exactly the same mutation was identified by Gill et al 8 in familial Coppock-like cataract. Here, we report the identical mutation in a large Indian family exhibiting sutural cataract with punctate and cerulean opacities. In addition, we present evidence that this mutation in the β-crystallin B2 gene is an independent event and most likely the result of gene conversion.
Identification and characterisation of the mutation
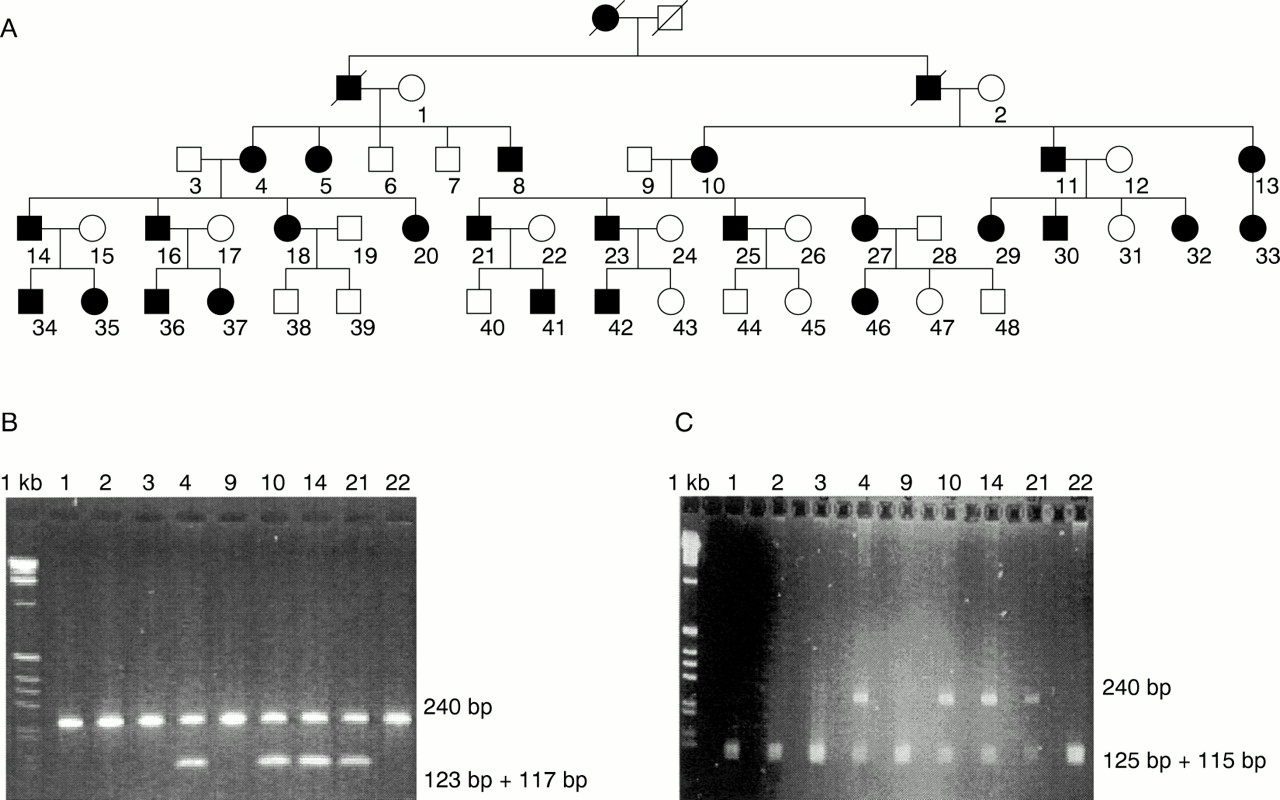
After obtaining informed consent, we performed linkage analysis in an Indian five generation family with 33 affected members, based on semi-automated genotyping with microsatellite markers from the Généthon linkage map; 48 members of this family, 25 of them affected, were selected for mapping (fig 1A). Assuming autosomal dominant inheritance with full penetrance and equal allele frequencies for each marker and using the LINKAGE program package, we calculated two point lod scores. After having excluded the autosomal dominant cataract loci on chromosomes 1, 2, 12, 13, 14, 16, 17, and 19, we detected linkage in our family to marker D22S315, with a lod score of Zmax = +8.500 at θmax = 0.05.

Pedigree of the family (A) and results of the restriction digestions of representative members. (B) CRYBB2 mutation Q155X creates a BfaI site. Restriction digestion with BfaI shows two unresolved digestion products of 117 bp and 123 bp in addition to the undigested 240 bp fragment in affected subjects. Unaffected persons show only an undigested 240 bp PCR product. Numbers above lanes correspond to individual numbers in the pedigree. 1 kb ladder is used as size standard. (C) CRYBB2 variant 483C→T destroys a MspI site. Restriction digestion with MspI shows the undigested 240 bp fragment in addition to two unresolved digestion products of 125 bp and 115 bp in affected subjects. Unaffected people show only two unresolved digestion products of 125 bp and 115 bp. Numbers above lanes correspond to subject numbers in the pedigree. 1 kb ladder is used as size standard.
This region on chromosome 22 harbours four β-crystallin genes,CRYBA4, CRYBB1,CRYBB2, CRYBB3, and the pseudogene CRYBP1.9 We amplified the translated exons 2-6 of theCRYBB2 gene by PCR as described previously.7 In addition, we designed primer sets for maximum discrimination between gene and pseudogene sequences: CRYBLg (5′-TGACCTTGTAGCTGGGCTTG-3′), CRYBLpsg (5′-TGACTTTGCAGCCAGGCTT G-3′), 596rg (5′-CACTGCATGTCGCGGAT ACG-3′), 596rpsg (5′-CCCTGCATGTCGT GGATGCA-3′). PCR products were purified with a Qiaquick PCR purification kit (Qiagen, Hilden, Germany) and sequenced directly using the Big-Dye-Terminator Cycle Sequencing Kit (PE Biosystems, Weiterstadt, Germany). Sequencing reactions were purified with a Dye-Ex Kit (Qiagen, Hilden, Germany) and run and analysed on ABI 310 and 377 sequencers (PE Biosystems, Weiterstadt, Germany).
Sequencing of exon 6 showed a C→T mutation at nucleotide position 475 (Q155X). This stop mutation truncates the protein by 51 residues and has previously been described.7 ,8 However, our sequencing of exon 6 showed an additional variant, a C→T substitution at nucleotide position 483. This silent polymorphism was found exclusively in patients. Since the mutation Q155X creates aBfaI restriction enzyme site and the variant 483C→T destroys an MspI site, PCR products were BfaI andMspI (New England Biolabs, Frankfurt, Germany) digested and separated on agarose gels. The results showed complete cosegregation of mutation, variant, and disease in our family (fig 1B, C). Q155X and 483C→T were not found on 180 chromosomes of normal Indian subjects, excluding either from being a frequent polymorphism.
To address the question whether the mutations in our Indian family and the American family of Litt et al 7 are derived from a single mutational event or represent recurrent mutations, we established the haplotypes at theCRYBB2 locus, based on closely linked microsatellite markers. Their order and genetic distance isTOP1P2 - 3 cM -CRYBB2 - 1 cM - D22S258.10 The haplotypes were 133-173-180 in the Indian and 165-171-180 in the American family, indicating that the mutation Q155X arose independently on chromosomes with different haplotypes. This is confirmed by the “normal” cytosine at position 483 in the patients of Littet al 7 and Gillet al,8 as can be seen from the published sequences.
Gene conversion
Altogether these data point to a hot spot of mutation in exon 6 of the CRYBB2 gene. Since the two sequence alterations in the Indian family, Q155X and 483C→T, are both cytosine to thymidine mutations, cytosine deamination as the mutational mechanism cannot be ruled out a priori. However, if one takes into account that the region harbours four closely related β-crystallin genes and a pseudogene (fig 2A), two other mutational mechanisms need to be considered, unequal crossing over and gene conversion.

The crystallin genes on chromosome 22. (A) Position and orientation of crystallin genes and physical distances between them.9. (B) Alignment of CRYBB2 exon 6 (italicised) with the homologous sequences of CRYBP1, CRYBB1, and CRYBB3. The closest similarity is observed between CRYBB2 and CRYBP1. The sequence of the CRYBB2 gene with the Q155X mutation (highlighted yellow) and the 483C→T variant (highlighted green) is identical to the CRYBP1 pseudogene sequence. Sequences that promote recombination events are underlined. We identified one hypervariable minisatellite GGGCAGGA(A/G)G with 1 bp mismatch and two chromosomal junction sequences ATGCAG with one mismatch each. Black and dark grey bars indicate sequence of probable and possible gene conversion, turquoise shows where gene conversion is excluded.
By alignment of the DNA sequences of the β-crystallin genes, it became clear that CRYBB2 is much more similar to the closely linked pseudogeneCRYBP1 than to the other β-crystallin genes (fig 2B). This finding suggests a recent duplication of a common precursor gene where one of the duplicated genes was maintained (CRYBB2) and the other one accumulated point mutations, turning it into a pseudogene (CRYBP1). The physical distance of 228 kb between CRYBB2 exon 6 and the homologous sequence in the pseudogene CRYBP1 (fig2A) is compatible with both unequal crossing over and gene conversion. To our surprise, the alignment of the crystallin gene sequences showed that the alterations Q155X and 483C→T in theCRYBB2 mutant are identical to the “normal” sequence in the pseudogene (fig 2B). Thus, the two alterations, Q155X and 483C→T, define a fragment of at least 9 bp of pseudogene-like sequence in the CRYBB2 gene. These 9 bp are flanked upstream by 28 bp and downstream by 67 bp where the gene and pseudogene are identical (fig 2B). At the nucleotides 29 bp upstream and 68 bp downstream that differ between gene and pseudogene, the patients showed homozygosity for theCRYBB2 sequence. Therefore, the mutant allele carries more than 9 bp but less than 104 bp of pseudogene-like sequence (fig 2B).
In principle, recombinational events resulting from unequal crossing over or gene conversion could explain this situation because the distance of 228 kb between gene and pseudogene (fig 2A) is compatible with both mechanisms. Unequal crossing over should lead to relatively large duplications or deletions. However, Southern blotting ofEcoRI digested genomic DNA with an exon 6 probe did not show any differences between affected and unaffected subjects (data not shown).
Thus, the most likely explanation is that gene conversion betweenCRYBB2 exon 6 and its homologousCRYBP1 sequence led to the pseudogene-like alteration in the CRYBB2 gene. This assumption is in agreement with the shortness (⩽104 bp) of the replaced fragment. It was estimated that gene conversions between human γ-globin genes are less than 300 bp in length11 and between human β-globin genes less than 451 bp.12 Our assumption of gene conversion is also supported by the presence of sequences that were described to promote gene conversion. Gene conversion is considered to be initiated at or near special sites.13 The critical region in our patients is flanked by the chromosomal junction sequence ATGCAG14 with one mismatch and also bears a hypervariable minisatellite GGGCAGGA(A/G)G15 with one mismatch (fig 2B). Putative gene conversion between highly homologous genes, as in this family, was previously described at several other loci, for example, the β-globin genes,16 the oxytocin vasopressin genes,17 or the steroid 21-hydroxylase gene and its pseudogene.18Summarising all our data, we conclude that gene conversion betweenCRYBB2 and its pseudogeneCRYBP1 is the most likely explanation for this mutation in exon 6 of the CRYBB2 gene.
Phenotypic variation and modifiers
There are now three families described that carry theCRYBB2 Q155X mutation. However, each of these shows a different phenotype. The clinical diagnosis in our Indian family is sutural cataract with punctate and cerulean opacities. The slit lamp examination (fig 3) showed prominent, dense, white opacification around the anterior and posterior Y sutures. The posterior Y sutures and the posterior pole of the lens were more severely affected than the anterior pole. It also showed greyish and bluish, sharply defined, elongated, spindle shaped, and oval punctate and cerulean opacities of various sizes arranged in lamellar form. The spots were bigger and more concentrated towards the peripheral layers. These did not delineate the embryonal or fetal nucleus. No pulverulent disc-like opacity was observed in the nuclear region. The sutural opacities appeared denser and whiter compared to the punctate and cerulean spots and were also more elongated and larger in size. Phenotypic variation with respect to the size and density of the sutural opacities as well as the number and position of punctate and cerulean spots was observed among the affected members. Some subjects showed severely affected sutures with dense white opacifications spreading along the secondary divisions of the Y sutures. In some affected subjects the spots were present only as a single layer in the cortex while in the others the spots occurred in concentric layers involving the whole cortex.

{kind=link}
{kind=link}
{kind=link}
3-D photograph of the eyes of a 9 year old patient through slit lamp microscope. In the centre of the lens there is a dense, white, sharply defined sutural opacity with two of its branches showing fish tail-like division towards the periphery. A large number of sharply defined, elongated, spindle shaped, and oval punctate opacities are directed radially. Similar presence and distribution of opacities are seen posteriorly which are much larger and denser than the anterior ones.
The phenotype of this Indian family, sutural cataract with punctate and cerulean opacities, differs from all other reported forms of cataract. The American family of Litt et al 7 has pure cerulean cataract, and the Swiss family of Gill et al 8 shows Coppock-like cataract. The phenotype of our family overlaps with the American family, both showing cerulean opacities. However, the sutural cataract and the punctate opacities in our phenotype have not been reported in the American or Swiss families. Moreover, the prominent pulverulent central disc-like opacity involving the embryonal and fetal nucleus seen in the Swiss family is not present in the American or this Indian family. There is not even an overlap between the phenotype of the Swiss family and the other two families.
Hence, we conclude that the Q155X mutation causes cataract formation but the distinct type of cataract depends on modifying genetic and epistatic factors. The influence of modifiers would make it impossible to infer the mutant gene from the cataract phenotype. Consequently, ADCC patients would need to be analysed for much more than only one ADCC gene.
Cis acting major modifiers could explain the considerable phenotypic variability between families. Minor modifying factors, acting in trans, could cause the phenotypic differences within families. The minor modifier of our family is obviously not linked to the Q155X mutation, since there is considerable clinical variability within our patients who share an identical haplotype. Interestingly, this excludes three further crystallin genes, CRYBA4,CRYBB1, andCRYBB3 (fig 2A), from being the minor modifier. On the other hand, exactly these crystallin genes are candidates for being the major modifier.
Acknowledgments
This work was supported by the German Ministry for Science and Technology and the Department of Biotechnology, Government of India. We thank the patients and their relatives for their cooperation and Michael Litt, Portland and Irene H Maumenee, Baltimore for DNA samples of the American family.