Article Text

Statistics from Altmetric.com
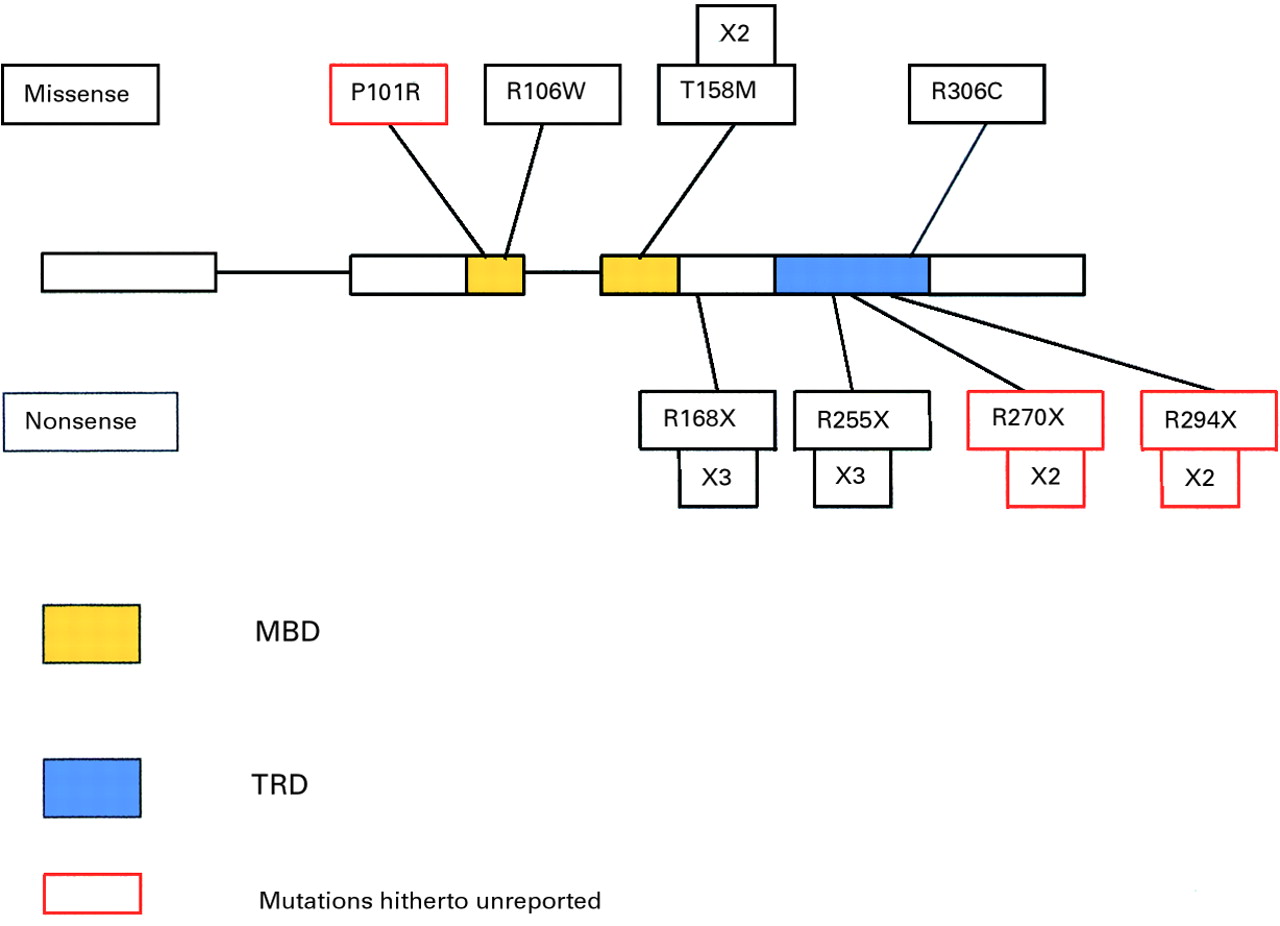
Editor—Rett syndrome is a severe, progressive, neurodevelopmental disorder which almost exclusively affects females. At first the affected girls appear to develop normally but after a year to 18 months they begin to deteriorate. Not only do they fail to progress but they lose skills already learnt until finally they have severe developmental delay with dementia and autistic behaviour, an apraxic gait, breathing dysfunction, and stereotyped hand movements, such as excessive hand wringing. Lost skills are not regained.1 The disease, which affects ∼1 in 10 000 girls, accounts for about 10% of profound handicap in females. More than 95% of cases are sporadic leading to the assumption that the syndrome must be the result of an X linked dominant gene with male lethality. Thomas2 has also suggested that the lack of males with the syndrome could be accounted for by the increased rate of de novo germline mutations in males. This would imply that affected females arise as a consequence of de novo mutations in their fathers. Marked skewing of X inactivation has not been detected in sporadic cases of the syndrome either in affected girls or in their mothers,3 but in one familial case the mother of three affected girls was found to have >95% skewing of inactivation in favour of the normal chromosome remaining preferentially active.4 The few available familial cases allowed the gene to be mapped to Xq285 6 and in 1999 Amiret al 7 reported that mutations in the MECP2 gene, located in Xq28, were associated with Rett syndrome in 5/21 of de novo cases. Amiret al 7 and Wanet al 8 reported a total of 10 mutations in the MECP2 gene of which five were missense and five were nonsense mutations. Four out of the five missense mutations were located in the highly conserved methyl binding domain (MBD), the fifth being in the equally highly conserved transcription repression domain (TRD) of the gene. The nonsense mutations, causing truncation of the protein product, were located both within and between these two functional domains. Wanet al 8 also found that certain of the truncating mutations were hot spots, the R168X mutation in particular being detected seven times. Because of this, despite the identification of equal numbers of individual missense and nonsense mutations, out of a total of 18 mutations detected, 12 were found to lead to a truncated product. Several other incidences of multiple recurrence were also detected, indicating the presence of further hot spots which all involved C→T transitions at CpG dinucleotides.
We report a mutation analysis of theMECP2 gene undertaken on a further 40 patients with Rett syndrome.
All of the girls entering the study had a clinical diagnosis of Rett syndrome. There were 27 singleton girls of European extraction, three Asian girls with consanguineous parents, and 10 girls from a further five British families.
DNA was extracted by standard methods from blood lymphocytes obtained from the girls diagnosed with Rett syndrome and from their families. The DNA was subjected to SSCP analysis using the primer sets published by Amir et al.7 The eight sets of primers covered all 3 exons of the MECP2gene but neither the 5′ untranslated region nor the very large 3′ untranslated region were investigated.
All band shifts detected on SSCP gels were sequenced by standard methods using an ABI 377 automatic sequencer. Both forward and reverse primers were used for sequencing PCR products with fluorescent dye terminators. The results were then compared to theMECP2 reference sequences published in the Gene Bank, X99686 for the transcribed and AF030876 for the complete gene sequence.
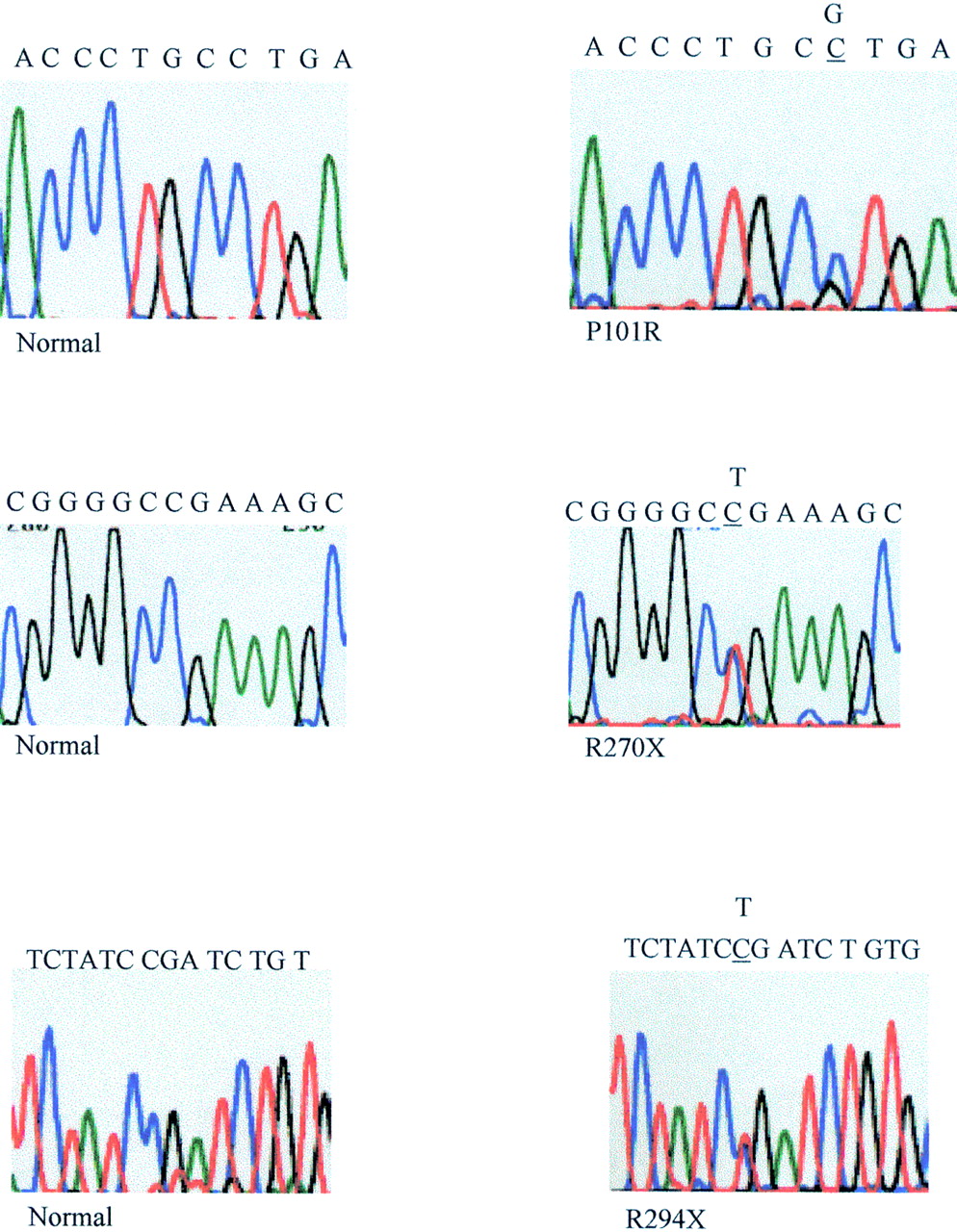
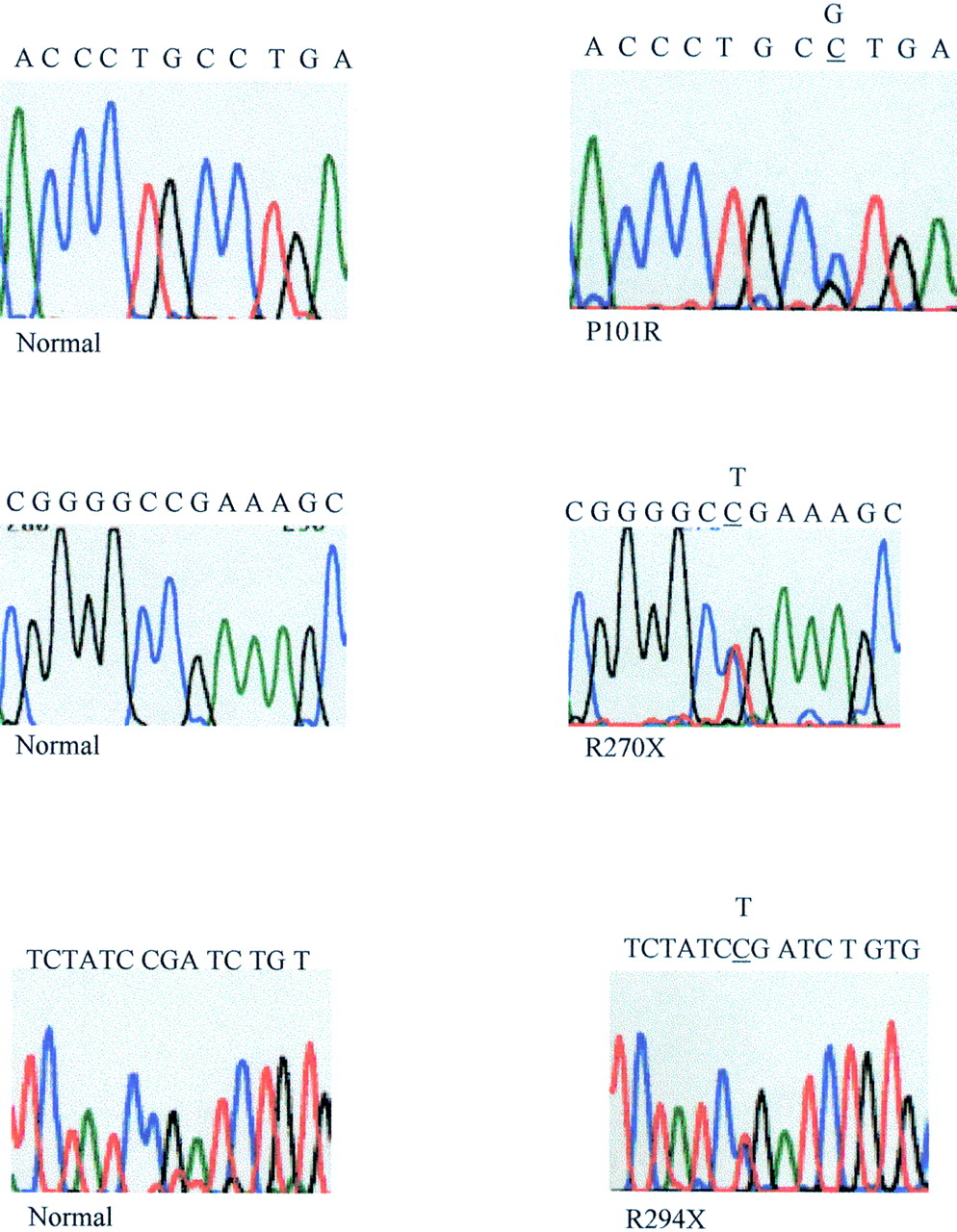
A total of 15 probands were found to have a mutation in theMECP2 gene. Two of these girls also showed a variant and a further two girls had variants alone (table 1). Altogether, eight different disease causing mutations were found (fig1), three were detected twice and a further two were found on three different occasions. Our results confirmed the presence of hot spots for mutation as suggested by Wan et al,8 as the R168X mutation which they found on seven occasions was detected a further three times in our cohort and the R255X which they found to reoccur once was also detected a further three times. Of the eight mutations detected, five had been previously reported by Amir et al 7 or Wanet al,8 but three have not been reported previously (fig 2). Interestingly two of these were detected twice in our cohort of affected girls, R270X in two European girls and R294X in another European and in an Asian girl. R270X is located in exon 3 and changes a CGA arginine codon to TGA stop codon, and R294X is also located in exon 3 and again changes a CGA arginine codon to a TGA stop codon. The third hitherto unreported mutation in this series, P101R, substitutes a CGT arginine codon for a CCT proline codon. This mutation is located in the MBD in a position conserved between human, mouse, chicken, and Xenopus. It is one of the very few mutations reported so far which substitutes a G and not a T moiety for a C and does not involve a CpG dinucleotide.
Detection of mutations and variants in the MeCP2 gene in Rett syndrome patients

Mutations detected in the MECP2 gene in Rett syndrome.
{kind=link}
{kind=link}
Sequences of the new missense and premature termination of translation mutations and normal controls (only forward sequences are shown).
One of the four variants detected (E397K) had also been reported by Wanet al,8 whereas the others were either silent or found to be located within introns.
Mutations that altered specific restriction enzyme cleavage sites could be confirmed by performing restriction digests. Other family members were also studied by this method if possible. The R106W and T158M mutations could be confirmed using theNlaIII site which is generated, the R168X mutation creates an HphI site, the R270X mutation creates an HaeIII site, and the R306C mutation creates an HhaI site. No unaffected family members were found to carry a disease causing mutation in the MECP2 gene.
In the 27 affected ethnically European girls, eight different mutations and two variants were detected. However, in the five families studied, out of 10 affected girls, only one had a mutation in theMECP2 gene (T158M); her affected cousin did not have this mutation. Linkage analysis with microsatellite markers had previously shown that the two affected girls shared alleles at Xq28 inherited from their respective mothers who are sisters. It is possible that the second cousin has a different mutation in theMECP2 gene as yet undetected or that she has a different form of neurological impairment. Amiret al 7 had previously reported that a pair of half sisters both had the R106W mutation. It was suggested that their mother, who did not have the mutation, was a gonadal mosaic.
If only the singleton girls are considered, then 12/27 (44%) of girls with ethnically European parents and 2/3 girls with consanguineous Asian parents had mutations. Thus, mutations have been detected in different racial groups and in some cases they have been shared, including one of the truncating mutations previously unreported.
The pattern of mutations in the MECP2 gene that are associated with Rett syndrome indicates that they fall into two types, nonsense mutations located in the TRD and missense mutations found almost exclusively in the MBD. The truncating mutations are believed to prevent the association of the 5-methylcytosine/MeCP2 complex with sin3A and histone deacetylase, preventing the deacetylation of histones and, as a consequence, failing to repress transcription.8 The missense mutations in the MBD are likely to interfere with the binding of MeCP2 to 5-methylcytosine itself. This would suggest that individual mutations should have different effects upon the phenotype, since MeCP2 is ubiquitously expressed and is believed to be involved in the silencing of many different genes. Mutations in the MECP2 gene have already been associated with different phenotypes,8but many individual mutations in different areas of the MBD still lead to the specific phenotype of Rett syndrome and it remains to be determined just which genes are affected and to what degree.