Article Text

Statistics from Altmetric.com
Editor—Rett syndrome (RTT, MIM 312760) is a neurodevelopmental disorder characterised by normal early psychomotor development followed by a period of regression, the loss of acquired purposeful manual and speech skills, hand wringing, gait disturbance, and growth retardation.1 As RTT occurs exclusively in females and almost all patients with RTT are sporadic, it has been proposed that RTT is caused by an X linked dominant mutation with lethality in hemizygous males.1 Recently, DNA mutations in the methyl-CpG binding protein 2 gene (MECP2), mapped to Xq28, have been detected in some patients with RTT.2 3 We carried out a mutation analysis in 40 Japanese patients with RTT to confirm thatMECP2 is the gene responsible for RTT and to detect common mutations in MECP2.
All patients screened in this study were sporadic cases, 38 patients with clinically typical phenotypes of RTT and two patients with preserved speech variant of RTT.4 Genomic DNA was extracted from the peripheral blood of 40 patients with RTT, their parents, and 105 healthy Japanese women. Primer pairs for polymerase chain reaction (PCR) amplification, designed using the genomic sequence of MECP2 (Gen Bank accession number, MeCP2 locus, AF030876, AJ132917), and the sizes of the products are shown in table 1. PCR amplification was performed in a final volume of 25 μl with PCR buffer, dNTPs, Taq polymerase, and each primer set. PCR conditions were: initial denaturing at 94°C for three minutes followed by 35 cycles of denaturing at 94°C, annealing at 56°C, and extension at 72°C for one minute each, and final extension for five minutes. To detect sequence variations, the products of PCR were analysed by electrophoresis on 6.5% polyacrylamide gels containing 0-100% linearly increasing denaturing agents at 60°C using a Bio-RAD D GENETM system and by direct DNA sequencing.
PCR primer sets for amplifing exons of MECP2
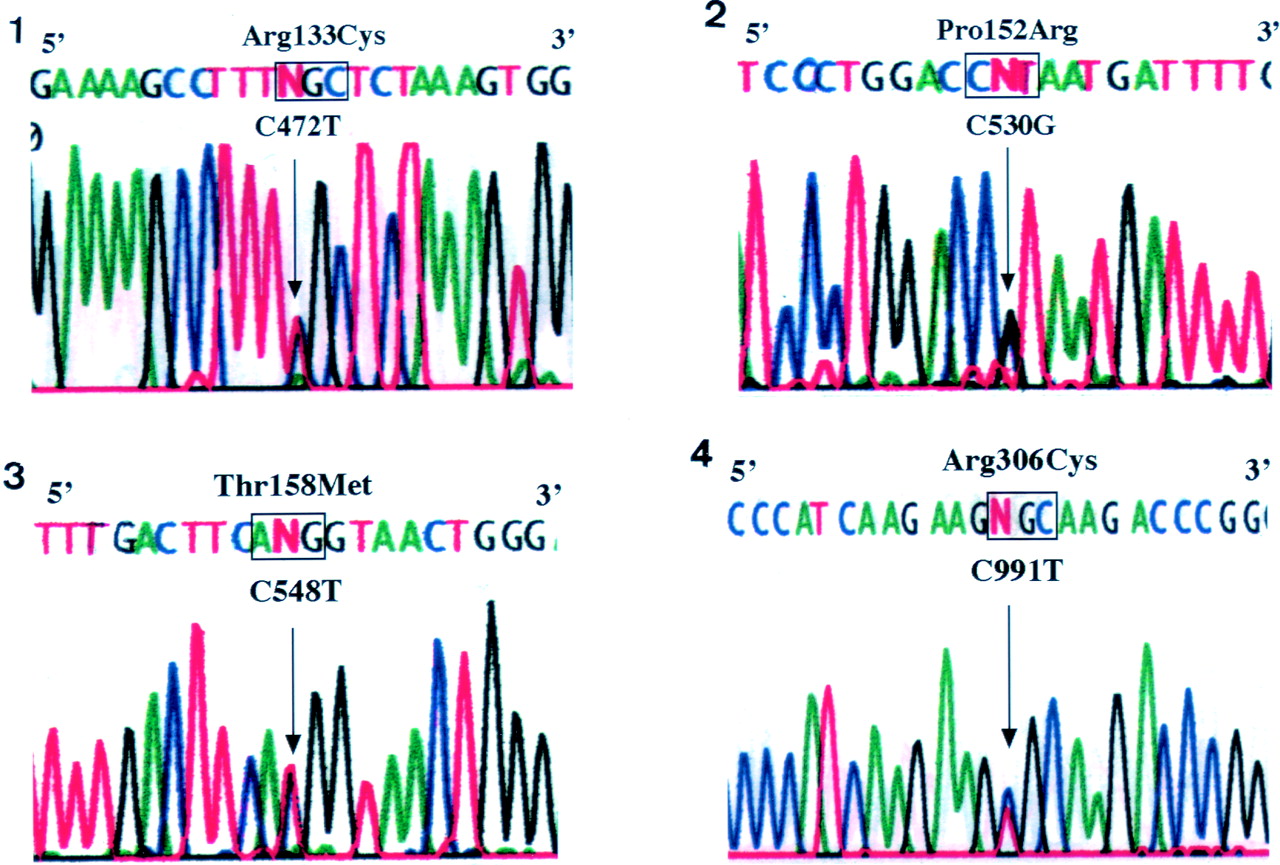
We found 15 different DNA mutations of MECP2in 36 (90%) of 40 patients with RTT (table 2). These mutations were not found in parents of patients or 105 healthy Japanese women, indicative of non-polymorphic variations. Three nonsense mutations, R168X, R270X, and R294X, were common in patients with RTT and have been identified in 12 (33.3%) of 36 patients with RTT (fig 1, table 2). In addition, four missense mutations, R133C, P152R, T58M, and R306C, were also detected in 16 cases with RTT (44.4%) (fig 2, table 2). Mutations with a nucleotide deletion resulting in a frameshift with a premature stop codon were detected in five patients with RTT. The clinical courses and symptoms in RTT patients with mutations ofMECP2 did not differ from those in four RTT patients who had no mutations detected in this study. Recently, Amiret al 2 and Wanet al 3 reported 10 mutations, five missense and five nonsense, in MECP2 in patients with RTT. Among the five missense mutations, four mutations (R106W, R133C, F155S, and T158M) were in a highly conservative methyl binding domain (MBD) of MECP2 and the R106W, R133C, and T158M mutations were also detected in patients in our study. In addition, three new missense mutations, L124F, S134C, and P152R, were detected in the MBD region of MECP2 in Japanese patients with RTT. Methyl CpG binding protein 2 (MeCP2), an abundant chromosomal protein with a high affinity for methyl CpG pairs, is a transcriptional repressor5 and is essential for embryonic development in mouse.6 Since MBD, which consists of 85 amino acids, is essential for chromosomal localisation of the protein as determined by a deletion study ofMECP2,5 these amino acid changes may decrease its binding affinity for chromatin.3
Mutations of MECP2 identified in patients with RTT

Nucleotide sequence of four common missense mutations of MECP2 in patients with RTT. (1) R133C mutation, (2) P152R mutation, (3) T158M mutation, (4) S306C mutation.

{kind=link}
{kind=link}
Nucleotide sequence of three common nonsense mutations of MECP2 in patients with RTT. (1) R168X mutation, (2) R270X mutation, (3) R294X mutation.
As a result of an extensive mutation search of CpG hotspots based on the MECP2 coding sequence, five R to X mutations, R168X, R255X, R270X, R294X, and R452X, were predicted by Wanet al.3 All mutations resulted from a C to T substitution in an Arg codon (CGA). Two nonsense mutations, R168X and R255X, were identified in a previous report.2 3 Five RTT patients in our study had the nonsense mutation R168X, resulting in a loss of the transcriptional repression domain (TRD) of MECP2. Furthermore, two similar nonsense mutations, R270X and R294X, were detected in three and four patients, respectively, with RTT (table 2). Twelve (33.3 %) of 36 patients with known DNA mutations ofMECP2 had one of these nonsense mutations in our series and had a truncated C-terminal half of MeCP2. The C-terminal half of the protein is needed for the efficient repression of transcription in vitro.7 These data suggest that a C to T substitution in an Arg codon (CGA) is the most common mutation inMECP2 in patients with RTT.
Seven mutations, R133C, P152R, T158M, S306C, R168X, R270X, and R294X, in exon 3 of MCEP2 were detected in 28 cases (77.8%) in our study. Six mutations resulted from a C to T transition and only one C to G transversion was observed in P152R. Thus, mutations in patients with RTT are restricted to some portions ofMECP2.
The clinical symptoms and their causes were slightly different in patients with different DNA mutations. Patients with the P152R mutation were severely handicapped; none of them can walk now. Patients with the R306C mutation had a mild form and all of them can walk and one patient can speak several significant words. In our study, the patients with R306C and R133C have the preserved speech variant. Further study of these links are necessary to confirm the genotype-phenotype correlation of RTT.
Amir et al 2 reported that five (23.8%) of 21 patients with RTT had DNA mutations inMECP2 in their preliminary study. Wanet al 3 detected DNA mutations of MECP2 in half of the patients with RTT. Furthermore, Zoghbi8 later found DNA mutations ofMECP2 in approximately 70% of patients with RTT. We found 15 different DNA mutations ofMECP2 in 36 (90%) of 40 patients with RTT, which is a much higher percentage than in the previous reports.2 3 8 We have determined the whole DNA sequence in encoding and splicing regions of MECP2 in patients who did not show heteroduplex DNA bands by the DGGE method. Thus, the rate of mutations of MECP2 in our study may increase compared with that in other studies.2 3 8 However, no mutations in the coding and splicing portions of MECP2 were detected in four of the patients with RTT screened so far. We have not analysed the 3′ untranslated region (3′-UTR) of MECP2.The long 3′-UTR of MECP2 is differently expressed in brain and other tissues, suggesting that both the primary sequence and the three dimensional structure of the 3′-UTR have essential roles in the post-transcriptional regulation ofMECP2 expression.6 Therefore, DNA mutations in the 3′-UTR of MECP2 may be responsible for the symptoms in patients with RTT. However, there is another possibility, that RTT is genetically heterogeneous and caused by other gene(s).
In conclusion, we have confirmed that mutations inMECP2 are responsible for RTT. Fifteen different mutations in MECP2 have been detected in 90% of patients with RTT and seven common mutations were defined. Early diagnosis of RTT is now possible in patients with neurodevelopmental problems using DNA analysis ofMECP2. Particular clinical symptoms were associated with DNA mutations in some patients with RTT and it may therefore be more heterogeneous than reported previously.