Article Text

Statistics from Altmetric.com
Editor—In recent years the increased resolution that can be obtained with GTL banded prometaphase chromosomes has led to the recognition of abnormalities involving small regions of the karyotype. Some of these abnormalities involve deletion or duplication of only one or two chromosomal bands and are associated with a sufficiently mild phenotype as to be directly inherited. Nevertheless, directly inherited duplication of visible chromosomal material is an uncommon category of chromosomal abnormality that has been reported for a small number of specific regions of the karyotype, including 2q11.2-q21.1,1 6p23-pter,26q22-q24,3 7p12-p13,4 8p23.1,59p22-p24,6 14q13-q22,7 15q12,8and 18p.9 Some of these duplications are without apparent phenotypic effect,5 8 while in other cases there are mild phenotypic abnormalities.1 2 Genomic imprinting has been shown to have an effect on the phenotypic expression of dup(15)(q12)8 and also dup(6)(q24),3 and is a point for consideration in other small duplications.
Tandem duplications occur when a second copy of a chromosomal region is inserted adjacent to the original region. They have been reported for a number of chromosomal segments,2 9 although not previously for band 10p14. Microduplication and microdeletion of chromosomal material is presumed to occur as a meiotic event following uneven crossing over between short sequences of highly similar DNA inserted at two close, but not contiguous sites along the chromosome.10 11 Tandem duplication is generally de novo with rare cases reported of direct inheritance.3-6 8 9To be balanced, an intrachromosomal duplication would have to be associated with chromosomal deletion of the specific region in the other homologue and to have occurred postzygotically. To our knowledge, this type of balanced chromosomal rearrangement has not been reported as a constitutional abnormality. We describe here a previously unreported small chromosomal duplication of band 10p14 segregating in a large pedigree with apparently direct inheritance in at least eight subjects, three of whom presented independently as index cases.
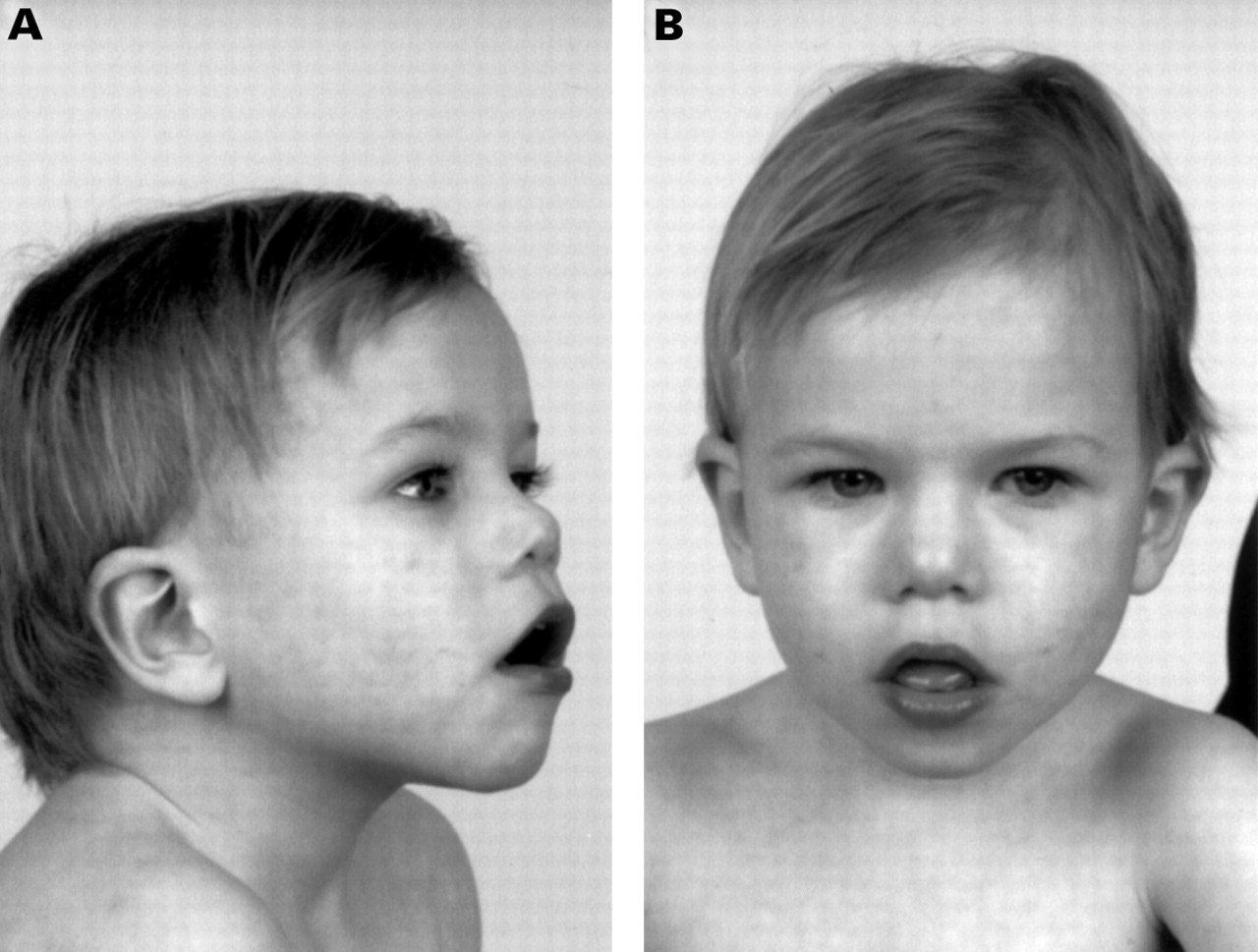
The pedigree of the family is shown in fig 1, with the three probands indicated. Proband 1 (IV.9 in fig 1, fig 2) was referred for chromosome studies at the age of 1 year 9 months with the clinical indications of intrauterine growth retardation, mild developmental delay, and small testes. A cleft palate had since been repaired. The child was in foster care and the foster parents reported the biological parents to be intellectually handicapped and the mother to have a very aggressive personality. There are two female sibs, both of whom are regarded as being intellectually normal, although neither has been seen by us. On review at 2 years 8 months, the child was small with height 88 cm (approximately 5th centile), weight 11 kg (3rd centile), and head circumference 47.7 cm (2 cm >3rd centile). He was being treated for seizures. He had no speech and no real vocalisation. Facial dysmorphism was definite but not marked, with the particular observations of a periorbital fullness, a flat nasal bridge with inner canthal folds, and a flattish midface (fig 2). The testes were abnormally small. There were bridged palmar creases on both hands and a degree of proximal implantation of the fourth toe on the right. Joint hypermobility was noted at the ankles.

Family pedigree. Subjects with the 10p duplication are shown as filled symbols, obligate heterozygotes as grey symbols, and subjects with mental defect of unknown cause who have not yet been cytogenetically studied are shown quarter filled. III.5, IV.6, and IV.7 have normal karyotypes. We understand III.7, IV.8, and IV.10 to be of normal intelligence.

IV.9 aged 2 years 8 months showing minor dysmorphism. Note the periorbital fullness, anteverted nares, and midfacial flatness with relative prognathism.
Proband 2 (IV.5, fig 1) was referred with developmental delay. On review at the age of 2 years 2 months, he had no speech and displayed hyperactive aggressive behaviour. There was no definite facial dysmorphism. His father (III.6) had an essentially normal facies and habitus, other than an upper spinal kyphosis. He could converse sensibly, intelligibly, and appropriately. He had had special schooling and had previously worked in a sheltered workshop, although at the time of interview he was in normal employment as a forklift driver. He was prone to anger, which he himself recognised as a problem. He expressed affection and concern for his son, having an insight into the difficulties that the chromosome abnormality had caused. However, he has since left the family, and the three children (IV.5-7) are now being brought up by their retarded mother and normal maternal grandparents.
Proband 3 (III.4, fig 3) was referred for chromosome analysis with the indications of intellectual impairment and short stature at the age of 14 years. On clinical examination at the age of 16, he was borderline microcephalic (head circumference 52.7 cm, 2nd centile). He had deep set eyes, a high nasal bridge with a broad nasal tip, flat midface, large mouth, and low set ears (fig 3). He had hypermobile joints. He would not allow examination of his genitalia. He was in special schooling and exhibited “boisterous” behaviour. His mother (II.2) had borderline mental abilities and had facial features similar to her son's (fig 3B). She had a nerve deafness. She reported her daughter III.1 to be mentally retarded. One grandchild (IV.4) is said by II.2 to be developmentally delayed, but neither he nor his mother (III.2) was available for study.

II.2 and III.4 at ages 16 and 47, respectively, showing only subtle dysmorphism. The clearest observation is of maxillary flatness.
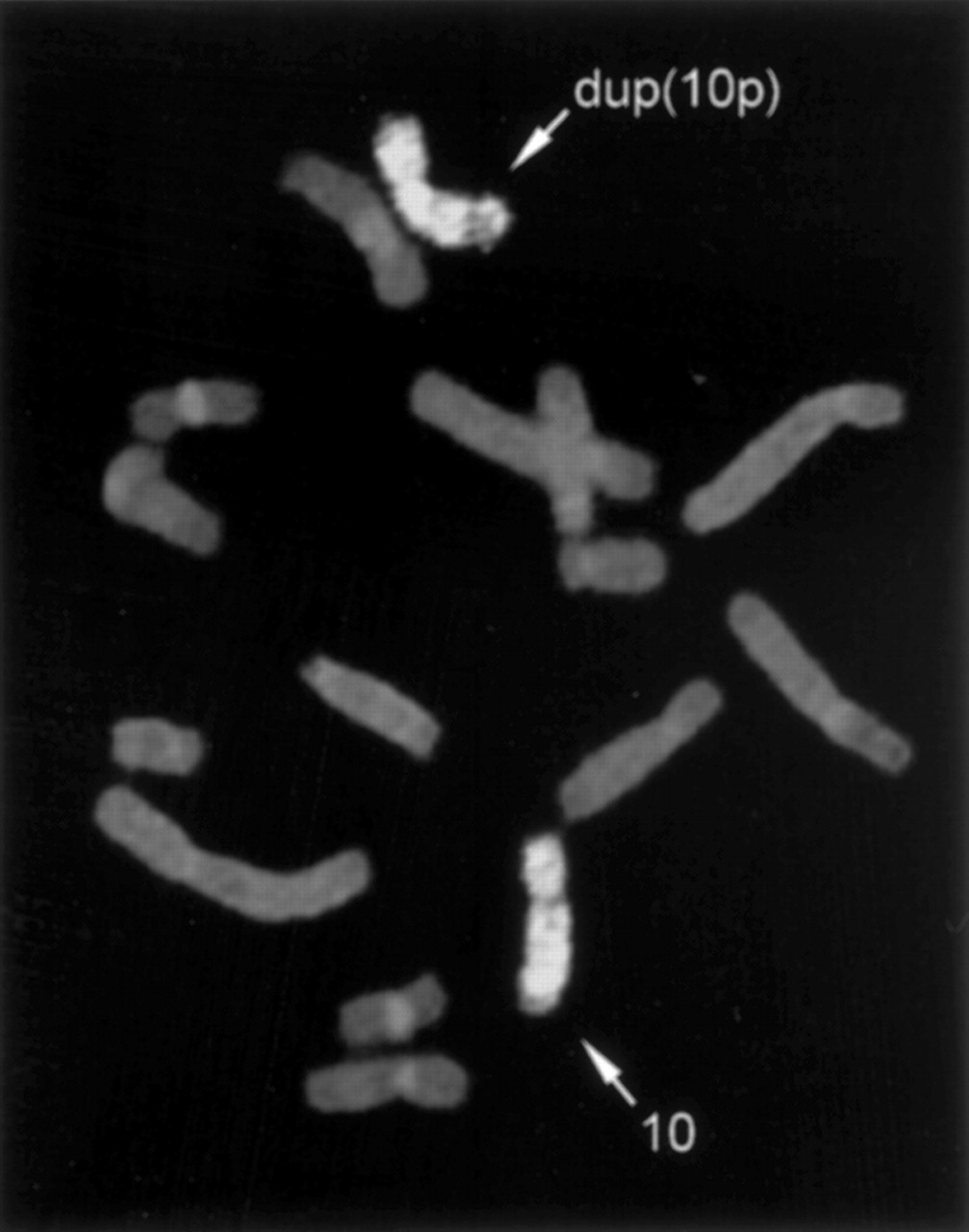
Metaphase cells were prepared for chromosome analysis from peripheral blood samples using standard techniques. Cells were synchronised using either excess thymidine or FudR with BrdU release. Chromosome analysis of the three probands showed in each an apparent duplication of band 10p14 (46,XY,dup(10)(p13p15)) (fig 4A, B, D). Chromosome studies on the parents of proband 2 had initially been interpreted to show mosaicism for the dup(10)(p14) in the father (III.6). Given the structure of the pedigree, as it subsequently came to light, and upon review of the cytogenetic material, his karyotype was reinterpreted as non-mosaic 46,XY,dup(10)(p13p15) (fig 4C). The duplication of band 10p14 could be observed in chromosomes around the 850 band level. In shorter chromosomes at 400-500 band levels, the duplication is observed as a more intense band than the normal 10p14 band present in the homologue, making it difficult to distinguish the duplication from banding variation. Chromosome studies on the mother of proband 3 also showed dup(10)(p13p15) (fig 4E). Her daughter had been referred for cytogenetic study in 1987 and a normal karyotype was reported at that time, but upon review of the archived slides her karyotype was reinterpreted as 46,XX,dup(10)(p13p15)mat. Chromosome painting was carried out using a chromosome 10 specific paint (Cambio, Cambridge, UK). Chromosome painting (fig 5) of metaphase cells from proband 1 (IV.9) using a chromosome 10 specific paint showed uniform hybridisation over both chromosomes 10 (with the exception of the heterochromatic pericentromeric region). This indicated that the rearrangement in the distal region of 10p was not the result of either interchromosomal insertion or translocation.

GTL banded partial karyotypes showing the normal chromosome 10 and the duplicated chromosome 10 (on the right) from (A) IV.9, (B) IV.5, (C) III.6, (D) III.4, (E) II.2, and (F) III.1.

Chromosome painting using a chromosome 10 specific paint showing uniform hybridisation to both the normal and duplicated chromosomes 10.
Probes previously mapped to 10p14 (JC2080) and 10p15 (JC2216) (obtained from Dr Jen-I Mao)12 were labelled with either biotin or digoxigenin by nick translation following the manufacturer's recommended method. Fluorescence in situ hybridisation (fig 6) on metaphase chromosomes from proband 3 (III.4) using probe JC2216 (10p15) showed hybridisation of comparable intensity to the terminal region of both the normal and duplicated chromosomes 10. In comparison, hybridisation with the probe JC2080 showed hybridisation to band 10p14 with increased intensity of signal present on the duplicated chromosome compared with the normal chromosome.

FISH studies on metaphase cells with duplication 10p14 using (A) probe JC2216 which maps to 10p15, showing hybridisation of equal intensity to both the normal and the duplicated chromosomes 10, and (B) probe JC2080 which maps to 10p14, showing increased hybridisation on the duplicated chromosome compared with the normal chromosome 10.
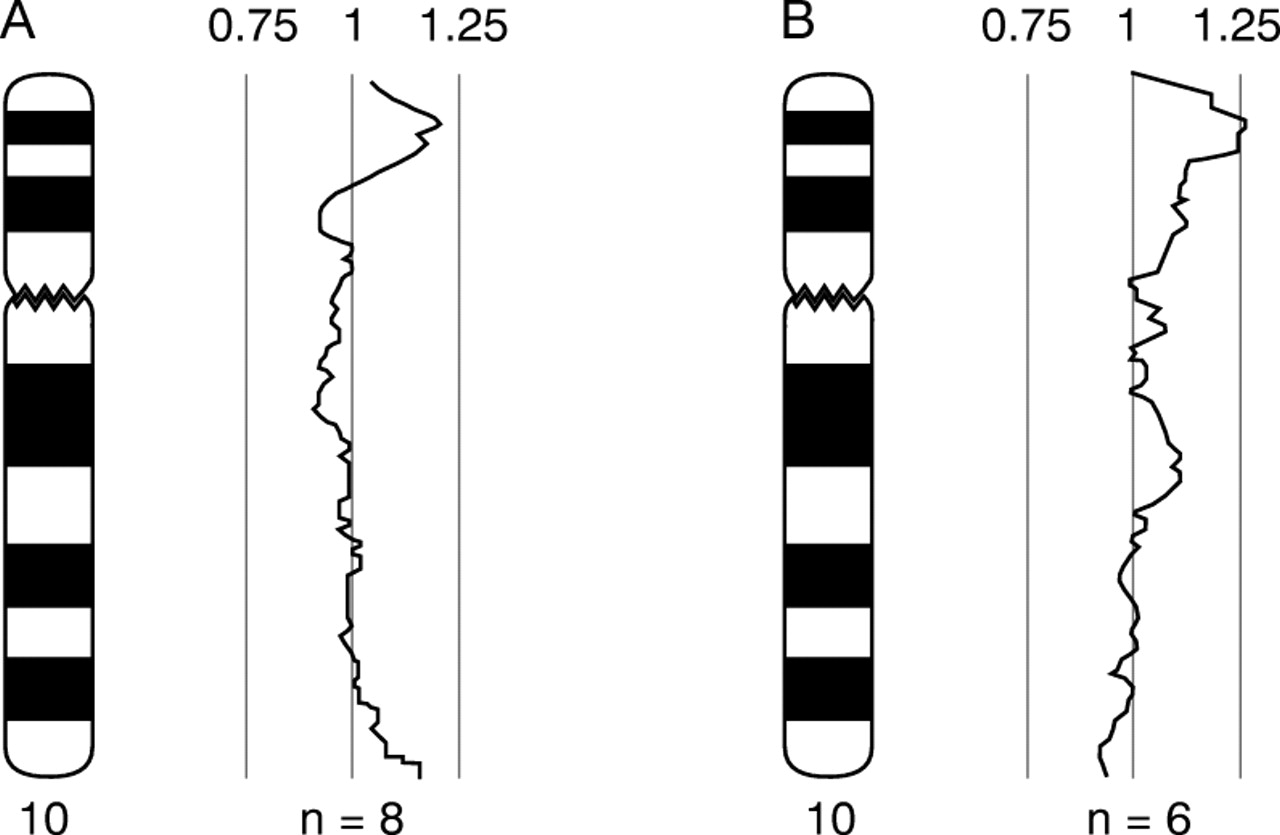
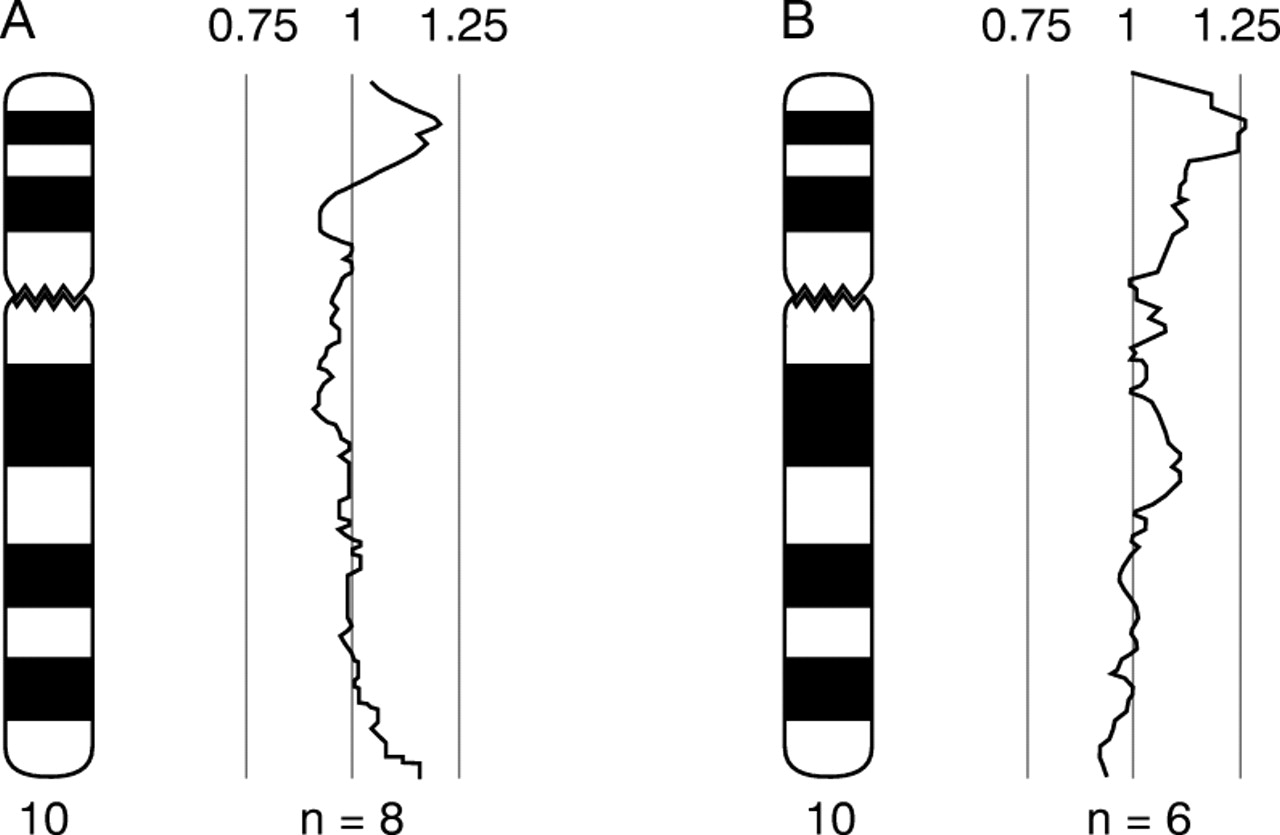
Comparative genomic hybridisation was performed using a modified protocol13 of the procedure described by Kallioniemiet al.14 Genomic DNA was extracted using the nucleon kit (Amersham) according to the manufacturer's instructions and directly labelled by nick translation using a Vysis kit according to the manufacturer's instructions. Test DNA was labelled with green fluorochrome and normal reference DNA was labelled with red fluorochrome. DNA samples obtained from proband 1 (IV.9) and from proband 2 (IV.5) were used in CGH experiments. For both samples, deviation of the profile towards the right occurred in the region of 10p14, indicating extra copies of DNA sequence in the test samples for this region (fig 7A).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CGH profile for chromosome 10 obtained from hybridisation of labelled genomic test DNA hybridised together with normal female genomic DNA. (A) Test DNA obtained from IV.9. (B) Test DNA obtained from IV.5 (n=number of chromosomes included in the profile). Deviation of the profile towards the right is consistent with extra DNA sequence copy number present in the test DNA compared with the reference DNA in the distal short arm region of chromosome 10.
Thus, we have described a chromosomal duplication of the band 10p14 that was ascertained independently during routine clinical cytogenetic analysis of GTL banded prometaphase chromosomes in three subjects, referred because of mental retardation. In two cases the abnormality was shown by cytogenetic studies to be directly inherited from a parent who also showed similar clinical features to those seen in the child. Review of the genetic files of the three probands showed that they all belonged to a single kindred (fig 1). Although some family members interviewed were aware of mental retardation and behavioural problems elsewhere in the family, they did not know of the chromosome abnormality. There had been considerable family dysfunction, and some of the presumed unaffected family members have been at pains to avoid contact with their relatives.
Chromosome painting indicated that the abnormality in the distal region of 10p involved only chromosome 10 material, with neither insertion nor translocation of chromosomal material from another chromosome. CGH studies on DNA from two of the probands showed that there was increased copy number for the DNA in the distal region of 10p. This confirmed the interpretation made on the basis of the GTL banding of prometaphase chromosomes, namely, that the chromosome rearrangement is a tandem duplication of band 10p14 with breakpoints in 10p13 and 10p15. FISH studies using probes mapped to bands 10p15 and 10p14 were confirmatory.
In terms of dysmorphology, the phenotype associated with this karyotype is mild, shading into apparent normality in case III.6. Hyperactive, impulsive, and intemperate behaviour is common and cognitive impairment is universal. A number of examples of 10p trisomy are on record,15 but to our knowledge only two show duplication for a very similar region. Stone et al 16 described dup(10p14→10pter) in a father and two daughters, and Benzacken et al 15 reported dup(10) (p14→pter) resulting from a de novo unbalanced translocation. This is a larger region than the duplicated segment in the present kindred, but there is some phenotypic overlap. All have had mental defect. In respect of physical attributes, midfacial hypoplasia, epicanthic folds, and anteverted nares are in common in our cases and in those of Stone et al.16 Optic nerve defects, subtle or major, were described in the three affected subjects in the family of Stoneet al 16 and agenesis of the corpus callosum was shown in the patient of Benzackenet al.15 These defects have not been specifically sought in our cases.
The presumed parent in generation I who has apparently transmitted the duplication to at least two offspring (II.2 and, inferentially, II.5) may have been a non-mosaic carrier of the duplication, although given family reports that I.1 and I.2 were both of normal intelligence, the possibility of mosaicism remains an open question. All other obligate carriers in this kindred must have duplication of 10p14 in non-mosaic state, inherited by direct transmission. Segregation of the duplication from a parent to an affected offspring is proven in 8/21 meiotic segregations, with at least two segregations uncertain. There is no obvious selection against the duplication, and the segregation pattern is comparable to that of an autosomal dominant condition. This suggests that the duplication does not interfere with meiotic pairing and segregation, and that in utero viability is little, if at all, compromised. Fertility is apparently not affected, albeit there is the observation of small testes in one child at the age of 2 years 8 months. The duplication has been transmitted both maternally and paternally, with no clear distinction of phenotypic patterns in the two classes of affected offspring, arguing against an imprinting effect. While direct transmission of chromosomal duplications (and deletions) is well described, this is the first report of a cytogenetically detectable chromosome duplication associated with phenotypic abnormality segregating through as many as four generations of one family. This genetic abnormality has been the cause of an extraordinary amount of inherited mental deficiency and psychopathology in the family.
Acknowledgments
Patients III.6 and IV.5 were referred by Dr I J Skelton, patient III.4 by Dr S Anderson, and patient IV.9 by Dr H Zehnwirth.